

Abstract
We studied the molecular evolution of the haemagglutinin (H) gene (full length) in all genotypes (24 genotypes, 297 strains) of measles virus (MeV). The gene’s evolutionary timescale was estimated by the Bayesian Markov chain Monte Carlo (MCMC) method. We also analysed positive selection sites. The MCMC tree indicated that the MeV H gene diverged from the rinderpest virus (same genus) about 250 years ago and that 24 MeV genotypes formed 3 lineages dating back to a 1915 ancestor (95% highest posterior density [HPD] 1882–1941) with relatively rapid evolution (mean rate: 9.02 × 10−4 substitutions/site/year). The 3 lineages diverged in 1915 (lineage 1, 95% HPD 1882–1941), 1954 (lineage 2, 95% HPD 1937–1969), and 1940 (lineage 3, 95% HPD 1927–1952). These 24 genotypes may have diverged and emerged between the 1940s and 1990s. Selective pressure analysis identified many negative selection sites on the H protein but only a few positive selection sites, suggesting strongly operated structural and/or functional constraint of changes on the H protein. Based on the molecular evolution of H gene, an ancestor MeV of the 24 genotypes emerged about 100 years ago and the structure of H protein has been well conserved.
Measles virus (MeV), which belongs to the genus Morbillivirus, the family Paramyxoviridae, causes measles, a highly contagious human disease manifesting as high fever, rash, respiratory, and conjunctivitis symptoms1. Severe complications may lead to pneumonia and encephalitis1.
Due to widespread measles immunisation programmes, the number of measles patients in many countries has been drastically reduced2. However, about 145,700 cases were estimated in 20132. The World Health Organization (WHO) is focusing on measles as an eliminative disease, and all six WHO regions have set elimination target years2.
To date, 24 MeV genotypes have been confirmed1, and the prevalence of each is associated with geographical area3. For example, genotype D4 was predominant in European countries from 2010 to 2013, while genotypes D8 and D9 were mainly detected in South-East Asian countries4. In China, the great majority of isolates are genotype H1 strains. In addition, genotype B3 is mainly detected in African areas4. Thus, although not well understood, the evolution of each MeV genotype differs by area.
The MeV genome encodes two major antigenic proteins, the haemagglutinin (H) and fusion (F) proteins1. The H protein regulates viral adsorption and entry, and that of the vaccine strains shows haemagglutination activity in erythrocytes from African green monkeys1,5. The neutralising antibodies against H protein are protective against MeV infection6. Detailed structures of the H protein and the epitopes for the neutralising antibodies have been reported7,8,9,10,11,12,13,14,15,16. The amino acid substitutions in H proteins may lead to antigenic changes10,17 related to the efficacy and development of the vaccine10.
Antigenic changes may occur through positive selection pressures in the host18. For example, frequent amino acid substitutions resulting from selective immune pressure in the host induce antigenic changes in the attachment glycoprotein (G) of respiratory syncytial virus (RSV)19,20. These changes are strongly linked to re-infections21. On the other hand, our previous report suggested that minor antigenic changes in the H protein are found in some MeV genotypes, such as D3, D5, D9, and H122. However, it is not known if these changes occur in other genotypes of MeV H protein.
Based on these studies, the molecular evolution of the H gene of MeV and changes in its antigenicity may be strongly linked. It is essential to be able to predict the antigenic changes of the various MeV genotypes to gain a better understanding of the efficacy of the vaccines. Therefore, we conducted a detailed genetic analysis of the H gene and predicted changes in the structure of H protein to gain an understanding of the evolution of the gene in all MeV genotypes.
Results
Phylogenetic analysis using the Bayesian Markov chain Monte Carlo (MCMC) and maximum likelihood (ML) methods on the full H gene of all measles virus genotypes and rinderpest virus
We used 297 strains and constructed the phylogenetic tree using the Bayesian MCMC method with the nucleotide sequences of the H gene (1854 nt) for 24 genotypes (A to H) of MeV and a strain of rinderpest virus (Fig. 1). Detailed data of the strains, including lineage, year of divergence, genotype, and rate of evolution, are presented in Table 1. The 95% highest posterior densities (HPDs) for each node of the phylogenetic tree are shown as grey bars in Fig. 1. We estimated the lineages of all genotype strains using a tree constructed by the ML method (Fig. 2). The MCMC tree estimated that the MeV diverged from the rinderpest virus, which belongs to the same genus, at approximately 1760 (95% HPD 1666–1838). These MeV genotype strains formed 3 major lineages (lineages 1 to 3, Fig. 2). Genotypes A, B, C, E, and F were classified as lineage 1. Lineage 2 contains genotypes G and H, and lineage 3 contains genotype D strains (D1–D11). The year of the first major division in the tree was estimated at approximately 1915 (95% HPD 1882–1941). The years of divergence of lineages 1 to 3 were 1915 (95% HPD 1882–1941), 1954 (95% HPD 1937–1969), and 1940 (95% HPD 1927–1952), respectively. All MeV genotypes emerged between the 1940s and 1990s. Details of the years of divergence of each lineage and genotype are shown in Table 1.
Figure 1. Phylogenetic tree of the haemagglutinin (H) gene constructed by the Bayesian Markov chain Monte Carlo method.

The Markov chain Monte Carlo tree was based on the full nucleotide sequence of the H gene (1854 nt) visualised in FigTree. Grey bars indicate 95% highest posterior density for the estimated year. The tree was estimated using an uncorrelated exponential relaxed clock model under an exponential growth model. The scale bar represents the unit of time (year). The reference strains of each genotype are shown in the tree. Common positively selected sites (aa 575) are indicated by a bold line.
Table 1. Year of divergence of each genotype.
| Genotypes | Year of divergence (95% HPD) | |
|---|---|---|
| 1 | A, B1, B2, B3, C1, C2, E, F | 1915 (1882–1941) |
| 2 | G1, G2, G3, H1, H2 | 1954 (1937–1969) |
| 3 | D1, D2, D3, D4, D5, D6, D7, D8, D9, D10, D11 | 1940 (1927–1952) |
| A | 1948 (1941–1952) | |
| B1 | 1974 (1966–1980) | |
| B2 | 1980 (1976–1982) | |
| B3 | 1979 (1972–1984) | |
| C1 | 1942 (1930–1950) | |
| C2 | 1969 (1959–1975) | |
| D1 | 1951 (1943–1957) | |
| D2 | 1979 (1971–1985) | |
| D3 | 1977 (1972–1982) | |
| D4 | 1974 (1968–1980) | |
| D5 | 1978 (1971–1985) | |
| D6 | 1973 (1957–1984) | |
| D7 | 1966 (1960–1971) | |
| D8 | 1983 (1975–1989) | |
| D9 | 1988 (1979–1994) | |
| D10 | 1992 (1983–1997) | |
| D11 | 1976 (1965–1986) | |
| E | 1955 (1937–1965) | |
| F | 1943 (1932–1950) | |
| G1 | 1965 (1951–1976) | |
| G2 | 1994 (1991–1995) | |
| G3 | 1993 (1990–1996) | |
| H1 | 1978 (1971–1984) | |
| H2 | 1986 (1977–1991) | |
HPD, highest posterior density.
Figure 2. Phylogenetic tree of H gene constructed by the maximum likelihood method.

Labels at the branch nodes show at least 70% bootstrap support. The scale bar shows nucleotide substitutions per site.
Next, the rate of molecular evolution of all genotypes was estimated from the tree as 9.02 × 10−4 substitutions/site/year (95% HPD 7.77 × 10−4–1.02 × 10−3). Divergence for each lineage and genotype was constant and the evolutionary rate was not substantial. The results suggested that MeV has 3 major lineages that uniquely evolved and formed each of the 24 genotypes.
Positive and negative selection sites in the deduced measles virus H protein
We analysed positive and negative selection sites in the strains using the single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL), and internal fixed effects likelihood (IFEL) methods. The dN/dS (non-synonymous rate/synonymous rate) values were calculated by these three methods. Detailed data are shown in Tables 2 and 3. First, 6 positive selection sites were found at aa 240, 476, 477, 481, 546, and 575 by the FEL method, 5 positive selection sites were found at aa 240, 282, 303, 315, and 575 by the IFEL method, and 2 positive selection sites were estimated at aa 546 and 575 by the SLAC method. Common sites of positive selection estimated by 3 methods were Q575. The global estimate of dN/dS was 0.25 (95% confidence interval 0.23–0.27) by the SLAC method. In addition, these common positive selection sites changed at 1940s–2000s (Fig. 1). Many negative selection sites were detected by each of the three methods (Table 3).
Table 2. Positive selection sites in the H gene.
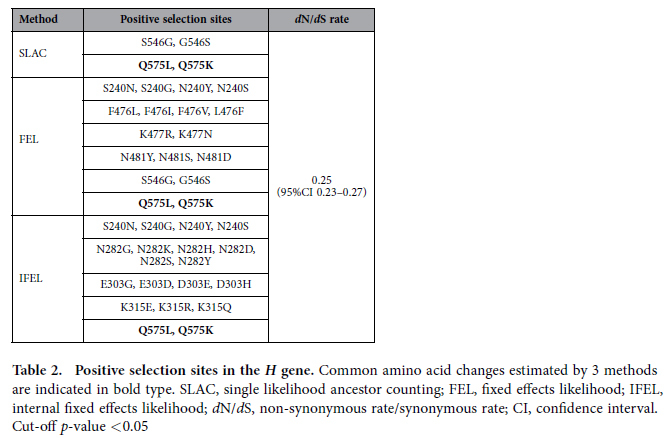
Table 3. Negative selection sites in the H gene.
| Method | Negative selection sites |
|---|---|
| SLAC | 181 sites |
| FEL | 242 sites |
| IFEL | 157 sites |
Cut-off p-value <0.05
A structure of a homotetramer (a dimer of dimers) of the H protein complexed with an MV receptor, signalling lymphocyte activation molecule (SLAM)11 was shown in Fig. 3. The individual H proteins in the homotetramer were shown in grey, light grey, purple, and light purple, and SLAMs were shown in cyan, as shown in previous papers14,15. Amino acid residues known to constitute a portion of an epitope were shown in blue, green, light green, yellow, orange, and red, as shown in previous papers14,15. The glutamine residue at position 575 (Q575) shown in magenta was also located at the surface of the H protein, but was unrelated to known antigenic epitopes (Fig. 3).
Figure 3. Mapping of the positive selection sites on the H protein structure.

An H protein homotetramer determined by Hashiguchi et al.11 (PDB3ALZ) is shown. (Left) Top view; (Right) bottom view. The individual H proteins in the tetramer are shown in grey, light grey, purple, and light purple, and SLAMs are shown in cyan. Amino acid residues known to constitute a portion of an epitope are shown in blue, green, light green, yellow, orange, and red. The glutamine residues at position 575 (Q575) are shown in magenta.
Phylodynamics of measles virus
We assessed the phylodynamics of the H gene of several MeV strains using Bayesian skyline plot (BSP) analyses (Fig. 4). Notably, from 1940 to the 1990s, the effective population sizes of the main strains, such as B3, D4, D8, D9, and H1, detected during the past 5 years (major genotypes)4 were smaller than those of other minor genotypes. However, the relative population sizes were reversed after 1995. The results suggested that major genotypes more adaptive than minor genotypes during around 15 years.
Figure 4. Bayesian skyline plot analysis of the measles virus H gene.
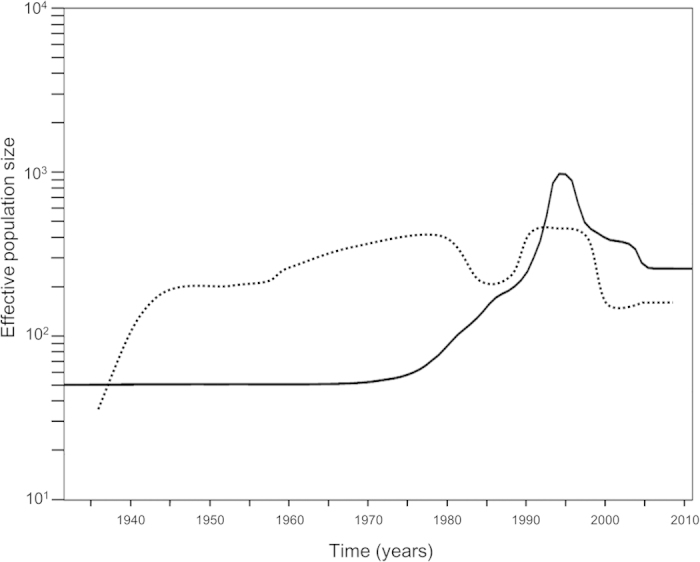
The Y-axis shows the effective population size and the X-axis shows generation time (year). Mean effective population size of major genotypes including B3, D4, D8, D9, and H1 are indicated by a solid line. Mean effective population size of minor genotypes such as B1, C1, E, and F are indicated by a dotted line.
Discussion
We studied the molecular evolution of the H gene in all known genotypes (24 genotypes, 297 strains) of MeV. First, we found that the MeV diverged from the common ancestors of rinderpest virus about 250 years ago during the 18th century. Twenty-four MeV genotypes were divided into 3 lineages dating back to a 1915 ancestor (95% HPD 1882–1941) based on the time-scaled evolutionary tree obtained using the Bayesian MCMC method. The three lineages diverged in 1915 (lineage 1, 95% HPD 1882–1941), 1954 (lineage 2, 95% HPD 1937–1969), and 1940 (lineage 3, 95% HPD 1927–1952). The 24 genotypes might have diverged and emerged between the 1940s and 1990s. Only a small number of positive selection sites were found, although many negative selection sites were identified. In addition, the recently effective population sizes of major genotypes (B3, D4, D8, D9, and H1)4 detected during the past 5 years were larger than those of minor genotypes. From the molecular evolution of the H gene, an ancestor MeV of these 24 genotypes emerged about 100 years ago, and the H protein structure has been well conserved. Such H protein conservation must be partially responsible for the effectiveness of the present vaccine.
Phylogenetic analysis in this study clearly showed the 3 lineages (Fig. 2). The strains belonging to lineage 1 form eight genotypes and are mainly detected in African, eastern Mediterranean, and western Pacific areas, while the strains of lineage 2 form five genotypes and are detected in Southeast Asian areas4. In contrast, the strains belonging to lineage 3 are detected across the world, including European, African, Asian, and American areas4. The strains of lineage 3 form many genotypes (D1 to D11), and genotypes D4, D8, and D9 are detected in many countries4. We also estimated the year of divergence of each MeV genotype, including rinderpest virus, using the Bayesian MCMC method. As a result, we found that MeV H gene diverged from a common ancestor of the rinderpest virus about 250 years ago (Fig. 1). After that, MeV uniquely evolved and formed 24 genotypes. Genotype C1 putatively emerged in 1942, the earliest year of divergence, while genotype G2 was the latest to emerge in 1994. In addition, Furuse et al., estimated that MeV diverged from rinderpest virus at around 11th to 12th centuries based on N (nucleoprotein) gene analysis using MCMC method23. Their and our results suggested that the genetic histories of the H and N genes might be distinct, although analysis of other genes may be needed to more fully describe the MeV genome.
Some prevalent strains, B3, D4, D8, D9, and H1, putatively emerged during the 1970s to 1980s. However, these genotypes have recently been detected as major genotypes. In contrast, some genotypes such as B1, E, F, G1, and G2 have rarely been detected the last 10 years4,24. These major genotypes showed wide genetic divergence in phylogenetic trees. In addition, BSP analysis suggested that the effective population sizes of the major genotypes were larger during the most recent 5 years than those of the minor genotypes (Fig. 4). These results suggest that the ancestor of these major genotypes might be more adaptive to humans, although we analysed only the H gene.
Previous reports have discussed the evolution rates of the H gene in MeV as ranging from 7.28 × 10−6 to 6 × 10−3 substitutions/site/year22,25,26,27. In the present study, we estimated that the mean rate of the molecular evolution of MeV H gene as 9.02 × 10−4 substitutions/site/year. While the present data were obtained from all 24 genotypes, other data were based on limited genotypes of MeV22,25,26,27. Thus, the variation among data from these studies may be partially attributed to the genotypes analysed.
We estimated positive and negative selection sites in the gene by SLAC, FEL, and IFEL methods. Positive selection imparts a survival advantage under the selective constraints that confront the viral population18. Negative selection plays an important role in maintaining the long-term stability of biological structures by removing deleterious mutations18,28. In this study, some positive selection sites were found (Table 2). A previous report showed that there are 14 positive selection sites in 50 strains of the H gene in various genotypes29. In addition, our previous report showed that the prevalent Asian genotypes, D3, D5, D9, and H1, have 8 positive selection sites estimated by FEL or IFEL methods22. Of these findings, amino acid positions corresponding to aa 476 and aa 575 are compatible with our results. In the previous report, aa 463–477 involving aa 476, and aa 561–575 involving aa 575, react with human sera for the neutralisation of MeV H protein22. Furthermore, aa 463–477 is a possible candidate for the binding site of the CD46 receptor22. The amino acid change at positive selection site 575 may confer an advantage to MeV transmission. However, the residue at position 575 was unrelated to the known effective neutralising epitopes. Therefore, an amino acid substitution at this position may not substantially affect the efficacy of humoral immunity against MeV. Nevertheless, only a small number of positive selection sites were identified for the MeV H protein, and there is little evidence to show that amino acid substitutions on the epitopes impart substantially better fitness to MeV in vivo. These observations could provide a rationale for the high level of efficacy of currently used MeV vaccines against all circulating MeV strains.
Many negative selection sites were found among the strains (Table 3). In general, negative selection is responsible for the prevention of functional deterioration of the viral proteins18. For example, negative selection sites in the neutralisation epitopes of polioviruses may be involved in receptor recognition and in the formation of altered particles30. Although the roles of these sites in MeV H protein are not exactly known, it is possible that these substitutions are involved in preventing the functional deterioration of major antigen (H protein)14,15.
In conclusion, the results suggest that the 24 MeV genotypes were generated over approximately 90 years and that the H protein was well conserved. This protein is an essential antigen of MeV. Thus, analysis of the biological properties of MeV H protein is required through the virus surveillance system. These studies will allow us to estimate the molecular evolution of MeV and give a better understanding of the aetiology of MeV.
Materials and methods
Strains and alignments
All collected sequence data were available in the DDBJ/GenBank database. We gathered a comprehensive collection of the full-length coding sequences for H gene of MeV. Sequence alignment was performed by CLUSTAL W31. Strains with 100% identity were excluded from the dataset. Furthermore, rinderpest virus (RBOK strain, Genbank accession No. Z30697) was added to the dataset. Accordingly, 297 strains of MeV and 1 strain of rinderpest virus were used in this study. The nucleotide sequences corresponded to positions 1–1854 of the H gene in strain MVi/Bethesda.Maryland.USA/54 (GenBank accession No. M81895). The detailed data are shown in Supplementary Table S1.
Phylogenetic analysis using the Bayesian Markov chain Monte Carlo method
To analyse the time-scaled phylogeny and evolutionary rate of H gene, we used the MCMC method in BEAST v1.7.532. The best nucleotide substitution model was selected using KAKUSAN433. The clock model and demographic model were compared by calculating Akaike’s information criterion through MCMC (AICM)34,35, for each model using Tracer v1.636. In this study, we tested 4 clock models: a strict clock, an uncorrelated lognormal relaxed clock, an uncorrelated exponential relaxed clock, and a random local clock37. Two demographic models, constant size model and exponential growth model, were also compared using AICM calculation34,35. We employed the model with the lowest AICM value. As a result, the datasets were analysed using a GTR-Γ model under an uncorrelated exponential relaxed clock with an exponential growth model. The data of each model are shown in Table S2. The MCMC chains were run for 75,000,000 steps and sampled every 10,000 steps. Convergence was assessed from the effective sample size (ESS) after a 10% burn-in using Tracer v1.636. ESS values above 200 were accepted. Three BEAST runs were combined using LogCombiner (available in BEAST package) following a 10% burn-in from each file. Uncertainty in the estimates was indicated by the 95% HPD intervals. The maximum clade credibility tree was generated by Tree Annotator v 1.7.4 after a 10% burn-in. The phylogenetic tree was viewed in FigTree v1.3.129. The evolutionary rates of each lineage were also estimated as described.
Phylogenetic analysis by the maximum likelihood methods
The phylogenetic tree of all MeV genotypes was constructed using the ML method with PhyML3.038. The best substitution model was selected using KAKUSAN433, and we employed the GTR-Γ model. The reliability of the tree was estimated using 1000 bootstrap replications.
Selective pressure analysis
Selection pressure analysis of the H gene was performed using Datamonkey39. We calculated the dS and dN rates at every codon using the following 3 methods: SLAC, FEL, and IFEL. The SLAC and FEL methods detect sites under selection at external branches of the phylogenetic tree, while the IFEL method investigates sites along the internal branches. SLAC is more conservative than other methods and intensive for large alignments40. However, this method appears to underestimate the substitution rate40. In contrast, FEL and IFEL methods consider both synonymous and nonsynonymous rate variations and may be efficiently parallelized40. Thus, we used the 3 different methods to obtain an accurate estimate of positive selection sites in the present study19,41. Positive (dN > dS) and negative (dN < dS) selection sites were determined using a p-value of <0.05.
Mapping of positive selection sites on the H protein structure
To clarify the positions of substituted amino acids at the positive selected sites on the H protein, we mapped them on the H protein structure reported by Hashiguchi et al.11. Figures were produced using PyMOL (DeLano Scientific LLC, Palo Alto, CA, USA. http://www.pymol.org).
Bayesian Skyline Plot analysis
To assess the differences in phylodynamics among each genotype, a BSP was constructed using the BEAST program32,42. We compared the effective population sizes of prevalent types4,24 such as B3, D4, D8, D9, and H1 (major genotypes, 159 strains) with those of other minor genotypes (147 strains). A substitution model was selected as described above. The MCMC chains were run for 150,000,000 steps with sampling every 10,000 steps under the strict clock, GTR-Γ model.
Additional Information
How to cite this article: Kimura, H. et al. Molecular evolution of haemagglutinin (H) gene in measles virus. Sci. Rep. 5, 11648; doi: 10.1038/srep11648 (2015).
Supplementary Material
Acknowledgments
This work was partly supported by a Grant-in-Aid from the Japan Society for the Promotion of Science and for Research on Emerging and Re-emerging Infectious Diseases from the Japanese Ministry of Health, Labour and Welfare (H25-Shinko-Ippan-010) and Japan Agency for Medical Research and Development.
Footnotes
The authors declare no competing financial interests.
Author Contributions H.K., M.S. and M.T. designed the study. A.N., K.K., M.K., F.T. and T.S. contributed analytic tools. H.K., M.K., H.I., T.S., D.K., H.T. and K.S. analysed data. H.K., M.K., A.R., H.M. and M.T. wrote the paper.
References
- Griffin D. E. [Measles viruses.] Fields virology. 5th ed. Vol. 2. [Knipe D. M., Howley P. M. (ed.)] [1551–1585] (Lippincott Williams & Wilkins, Philadelphia, 2007). [Google Scholar]
- Global progress towards regional measles elimination, worldwide, 2000-2013. Wkly. Epidemiol. Rec. 89, 509–516 (2014). [PubMed] [Google Scholar]
- Rota P. A. et al. Global distribution of measles genotypes and measles molecular epidemiology. J. Infect. Dis. 204, S514–S523 (2011). [DOI] [PubMed] [Google Scholar]
- World Health Organization. Immunization, Vaccines and Biologicals - Measles Surveillance Data. Regional summary of reported measles cases. (2014) Available at: http://www.who.int/immunization/monitoring_surveillance/burden/vpd/surveillance_type/active/measles_monthlydata/en/index1.html (Accessed: 4th October 2014).
- Rosen L. Hemagglutination and hemagglutination-inhibition with measles virus. Virology. 13, 139–141 (1961). [DOI] [PubMed] [Google Scholar]
- de Swart R. L., Yüksel S. & Osterhaus A. D. Relative contributions of measles virus hemagglutinin- and fusion protein-specific serum antibodies to virus neutralization. J. Virol. 79, 11547–11551 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouche F. B., Ertl O. T. & Muller C. P. Neutralizing B cell response in measles. Viral Immunol. 15, 451–471 (2002). [DOI] [PubMed] [Google Scholar]
- Ertl O. T., Wenz D. C., Bouche F. B., Berbers G. A. & Muller C. P. Immunodominant domains of the Measles virus hemagglutinin protein eliciting a neutralizing human B cell response. Arch. Virol. 148, 2195–2206 (2003). [DOI] [PubMed] [Google Scholar]
- Fournier P. et al. Antibodies to a new linear site at the topographical or functional interface between the haemagglutinin and fusion proteins protect against measles encephalitis. J. Gen. Virol. 78, 1295–1302 (1997). [DOI] [PubMed] [Google Scholar]
- Hashiguchi T. et al. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. USA 104, 19535–19540 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi T. et al. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat. Struct. Mol. Biol. 18, 135–141 (2011). [DOI] [PubMed] [Google Scholar]
- Hu A., Sheshberadaran H., Norrby E. & Kövamees J. Molecular characterization of epitopes on the measles virus hemagglutinin protein. Virology. 192, 351–354 (1993). [DOI] [PubMed] [Google Scholar]
- Liebert U. G. et al. Antigenic determinants of measles virus hemagglutinin associated with neurovirulence. J. Virol. 68, 1486–1493 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara, M. et al. Functional and structural characterization of neutralizing epitopes of measles virus hemagglutinin protein. J. Virol. 87, 666–675 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara M. et al. The receptor-binding site of the measles virus hemagglutinin protein itself constitutes a conserved neutralizing epitope. J. Virol. 87, 3583–3586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler D. et al. Protection against measles virus encephalitis by monoclonal antibodies binding to a cystine loop domain of the H protein mimicked by peptides which are not recognized by maternal antibodies. J. Gen. Virol. 77, 2479–2489 (1996). [DOI] [PubMed] [Google Scholar]
- Massé N. et al. Measles virus (MV) hemagglutinin: evidence that attachment sites for MV receptors SLAM and CD46 overlap on the globular head. J. Virol. 78, 9051–9063 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E. [Virus evolution.] Fields virology. 5th ed. Vol. 1. [Knipe D. M., Howley P. M. (ed.)] [389–421] (Lippincott Williams & Wilkins, Philadelphia, 2007). [Google Scholar]
- Kushibuchi I. et al. Molecular evolution of attachment glycoprotein (G) gene in human respiratory syncytial virus detected in Japan 2008-2011. Infect. Genet. Evol. 18, 168–173 (2013). [DOI] [PubMed] [Google Scholar]
- Hirano E. et al. Molecular evolution of human respiratory syncytial virus attachment glycoprotein (G) gene of new genotype ON1 and ancestor NA1. Infect. Genet. Evol. 28, 183–191 (2014). [DOI] [PubMed] [Google Scholar]
- Yamaguchi M. et al. High frequency of repeated infections due to emerging genotypes of human respiratory syncytial viruses among children during eight successive epidemic seasons in Japan. J. Clin. Microbiol. 49, 1034–1040 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M. et al. Molecular evolution of hemagglutinin (H) gene in measles virus genotypes D3, D5, D9, and H1. PLoS One. 7, e50660 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse Y., Suzuki A. & Oshitani H. Origin of measles virus: divergence from rinderpest virus between the 11th and 12th centuries. Virol J. 7, 52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. MeaNS. Distribution of measles genotypes from Jan to Dec 2013. (2014) Available at: http://www.hpa-bioinformatics.org.uk/Measles/Public/Data_Mnt/who_map.php (Accessed: 3rd February 2014).
- Rima B. K. et al. Sequence divergence of measles virus haemagglutinin during natural evolution and adaptation to cell culture. J Gen Virol. 78, 97–106 (1997). [DOI] [PubMed] [Google Scholar]
- Xu S. et al. Genetic characterization of the hemagglutinin genes of wild-type measles virus circulating in china, 1993-2009. PLoS One. 8, e73374 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Alia M. A. et al. Measles virus genetic evolution throughout an imported epidemic outbreak in a highly vaccinated population. Virus Res. 196, 122–127 (2015). [DOI] [PubMed] [Google Scholar]
- Loewe L. Negative selection. Nature. Education. 1, 59 (2008). [Google Scholar]
- Woelk C. H., Jin L., Holmes E. C. & Brown D. W. Immune and artificial selection in the haemagglutinin (H) glycoprotein of measles virus. J. Gen. Virol. 82, 2463–2474 (2001). [DOI] [PubMed] [Google Scholar]
- Suzuki Y. Negative selection on neutralization epitopes of poliovirus surface proteins: implications for prediction of candidate epitopes for immunization. Gene. 328, 127–133 (2004). [DOI] [PubMed] [Google Scholar]
- Larkin M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- Drummond A. J. & Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe A. S. Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol. Ecol. Resour. 11, 914–921 (2011). [DOI] [PubMed] [Google Scholar]
- Suchard M. A., Weiss R. E. & Sinsheimer J. S. Bayesian selection of continuous-time Markov chain evolutionary models. Mol Biol Evol. 18, 1001–1013 (2001). [DOI] [PubMed] [Google Scholar]
- Kass R. E. & Raftery A. E. Bayes factors. J Am Stat Assoc. 90, 773–795 (1995). [Google Scholar]
- Rambaut A. & Drummond A. J. Tracer. (2013) Available at: http://tree.bio.ed.ac.uk/software/tracer (Accessed: 11th December 2014).
- Drummond A. J., Ho S. Y., Phillips M. J. & Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59, 307–321 (2010). [DOI] [PubMed] [Google Scholar]
- Pond S. L. & Frost S. D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 21, 2531–2533 (2005). [DOI] [PubMed] [Google Scholar]
- Pond S. L. & Frost S. D. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 22, 1208–1222 (2005). [DOI] [PubMed] [Google Scholar]
- Botosso V. F. et al. Positive selection results in frequent reversible amino acid replacements in the G protein gene of human respiratory syncytial virus. PLoS Pathog. 5, e1000254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A., Shapiro B. & Pybus O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 22, 1185–1192 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.