

Abstract
High-risk human papillomavirus (HPV)16/18 infection in the development of lung cancer has previously been identified, and fragile histidine triad (FHIT) loss and p53 mutation are frequently observed in the disease. However, the association between these factors has not been well studied. The present study aimed to further investigate the significance of HPV infection, FHIT loss and p53 mutations in the development of lung cancer and their possible associations. DNA was extracted from paraffin-embedded specimens from 88 cases of squamous cell carcinoma (SCC), 56 of adenocarcinoma (AC), 36 of small cell lung carcinoma (SCLC) and 110 non-cancer control cases of lung neoplasms. The prevalence of HPV infection was determined by polymerase chain reaction analysis, and FHIT loss and p53 mutations were detected by immunohistochemistry. The χ2, Fisher's exact and Pearson correlation tests were applied for statistical analysis. The results of the present study demonstrated that HPVL1 (the major capsid protein of HPV), HPV16 and HPV18 infection were more prevalent in the lung cancer samples compared with the non-cancer controls (all P<0.001). FHIT loss occurred more frequently in the lung cancer samples (44.44%) compared with the non-cancer controls (7.25%) (P<0.001). FHIT loss in the HPVL1-positive group was significantly increased compared with the HPVL1-negative group in the lung cancer cases and the non-cancer controls (P<0.05). In the lung cancer cases, the p53 mutation rates in the HPVL1- and HPV16/18-positive groups were significantly increased compared with the HPVL1- and HPV16/18-negative groups (P<0.05). In the 180 lung cancer cases, the coexistence rate of FHIT loss and a history of smoking was 38.33% (69/180; Pearson contingency coefficient of r=0.318; P<0.001). FHIT loss and p53 mutation exhibited a synergistic effect on HPV-associated lung cancer (Pearson contingency coefficient r=0.357, P<0.001). The present study demonstrated that FHIT loss may be important in the occurrence of lung cancer, particularly in lung SCCs. FHIT loss may therefore be used as an early indicator for lung cancer, particularly for patients with a history of smoking. HPV infection in lung tumorigenesis may, at least in part, be mediated through FHIT loss. FHIT loss and p53 mutation may coordinate together in the development of HPV-associated lung cancer, and accelerate the occurrence and development of lung cancer.
Keywords: lung cancer, human papillomavirus, FHIT loss, p53 mutation
Introduction
Human papillomavirus (HPV) is a double-stranded DNA virus, and is an established etiological agent that may lead to the development of cervical cancer following infection. In total, >100 different HPV genotypes have been identified. HPVL1 is the main structural and major capsid protein of HPV, which contains stable immunogenicity. Thus, HPVL1 detection can indicate HPV infection. High-risk HPV16 and HPV18 are the most common oncogenic types, leading to the development of cervical carcinomas and a variety of different types of reproductive system malignant tumor (1–4). Lung cancer is one of the most common cancers and the leading cause of cancer-associated mortality in the world. The pathogenesis of lung cancer is complex, and is considered to be a result of the interaction between environmental and genetic factors. Previous studies have demonstrated that high-risk HPV16/18 may be closely associated with lung cancer (5–9), but the mechanisms are not yet fully elucidated.
The fragile histidine triad (FHIT) gene is a candidate tumor suppressor gene located at chromosome 3p14.2 and encompassing the FRA3B common fragile site (the most active chromosome breakage site in the human genome). FHIT anomalies are associated with the occurrence and development of a variety of malignant tumors. When a tumor develops, tumor cell proliferation gradually increases, while FHIT protein expression decreases. A previous study of cervical squamous cell carcinomas (SCCs) demonstrated that the HPV16 integration point was between FHIT exon 3 and 5. During the process of HPV16 integration, it may induce the deletion of exons within FHIT leading to its inactivation (10–12). Although data has indicated that there may be an association between FHIT loss and HPV infection, the exact mechanism remains poorly understood. FHIT loss is a frequent event in HPV-positive lung cancer (13), indicating that FHIT loss may be significant in the occurrence and development of HPV-associated lung cancer. FHIT loss has been observed in the early stages of tumorigenesis and may be a potential marker of HPV-associated lung cancer.
Our previous study identified that mutations in p53 contributed to high-risk HPV16/18-infected lung cancer cases (14). The role of p53 is to safeguard the integrity of the genome by inducing cell cycle arrest or apoptosis in response to DNA damage (15). In HPV16/18-infected lung cancer cases, E6 is bound to the p53 tumor suppressor gene, and thereby induces p53 degradation or mutation (14). One hypothesis is that there is a correlation between high-risk HPV16/18 infection and FHIT loss and p53 mutation. However, further study has not been conducted to investigate this potential association in HPV-associated lung cancer. The role of HPV16/18 in the development of lung cancer has previously been investigated, and FHIT loss and p53 mutation have been frequently observed in the disease; however, the association between them is not well studied. The present study further investigated the significance of HPV infection, FHIT loss and p53 mutation in the development of lung cancer and their possible associations.
Materials and methods
Ethical statement
The study was approved by the Ethics Committee of Xi'an Jiaotong University (Xi'an, Shaanxi, China; no. 20120099). Written informed consent was obtained from each patient prior to participation in the study.
Patients and samples
A total of 290 samples, including 88 lung SCCs, 56 lung adenocarcinomas (AC), 36 lung small cell lung carcinomas (SCLC) and 110 non-cancer controls were collected from patients diagnosed in The First Affiliated Hospital of Medical School of Xi'an Jiaotong University. All cases were selected from the period between January 2012 and May 2014, and all specimens were classified by three experienced pathologists according to the World Health Organization classification.
The lung cancer samples included 85 undifferentiated or poorly-differentiated carcinomas, 57 moderately-differentiated carcinomas and 38 well-differentiated carcinomas. The samples were obtained from 53 female (29.44%) and 127 male (70.56%) individuals, with an age range between 30 and 80 years and a mean age of 51.56 years. A history of smoking was present in 124 cases (68.89%) and absent in 56 cases (31.11%).
The 110 non-cancer control samples included 38 bronchiectasis samples, 21 inflammatory pseudotumors, 33 pulmonary tuberculomas and 18 pulmonary cysts. The samples were from 18 female (16.36%) and 92 male (83.64%) individuals, with an age range of between 25 and 75 years and a mean age of 51.48 years.
Reagents
The rabbit anti-FHIT polyclonal antibody (primary antibody; dilution, 1:20; cat. no. PA5-32418; incubation, 4°C overnight) was produced by Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The Universal LSABTM+ immunohistochemical kit (cat. no. K0679), including the biotinylated goat anti-rabbit antibody (secondary antibody; dilution, 1:50; incubation, 37°C for 15 min) and enzyme-chain avidin complexes (incubation, 37°C for 15 min), was produced by Dako (Glostrup, Denmark).
DNA extraction
The 4% paraformaldehyde-fixed and paraffin-embedded tissue samples were cut into 10-µm slices and placed into 1.5-ml Eppendorf tubes as described previously (16). The specimens were then treated with 1 ml of xylene, followed by 1 ml of ethanol. Following centrifugation at 14,020 × g at 4°C for 5 min, the pellet, which contained the precipitated DNA, was resuspended in digestion buffer (50 mM Tris-Cl, pH 8.0; 1 mM EDTA, pH 8.0; 0.5% Tween-20) containing 200 µg of proteinase K (Invitrogen Life Technologies, Carlsbad, CA, USA) and incubated at 56°C overnight. After incubation, the solution was heated at 100°C for 10 min and centrifuged at 14,020 × g at 4°C for 10 min. An aliquot of the supernatant, containing the precipitated DNA, was stored at −20°C prior to use.
β-globin polymerase chain reaction (PCR)
The method for β-globin detection and evaluation was performed as previously described (6). PCO3/PCO4 (110 bp) primers (PCO3 primer, 5′-ACACAACTGTGTTCACTGC-3′; and PCO4 primer, 5′-CAACTTCATCCACGTTCACC-3′) were used as the internal positive control. The PCR conditions were as follows: Initial denaturation at 95°C for 4 min, 40 cycles with a cycling profile of 95°C for 1 min, 52°C for 1 min and 72°C for 2 min, and a final extension for 5 min at 72°C.
Quantitative PCR
The thermal conditions of amplification were as follows: Initial denaturation at 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 45–55°C for 30 sec and 72°C for 1 min, and a final extension at 72°C for 5 min. The primer sets were synthesized by Shaanxi Jintai Biological Engineering Co., Ltd., (Xi'an, China) and are presented in Table I.
Table I.
HPV primer sets and length.
| Primer | Sequence | Length, bp |
|---|---|---|
| HPVL1 | 5′-CGTCCCAGGGGATACTGATC-3′ | 450 |
| 5′-GCCCAGGGTCATAATAATGG-3′ | ||
| HPV16 | 5′-GAACAGCAATACAACAAACCCG-3′ | 315 |
| 5′-CCATGCATGATTACAGCTGG-3′ | ||
| HPV18 | 5′-TGCCAGAAACCGTTGAATCC-3′ | 152 |
HPV, human papillomavirus.
Negative and positive controls were designed for each experiment. Sterilized water was used as the negative control and a HPV-infected condyloma acuminatum specimen was used as the HPV general positive control. The HPV16- and HPV18-positive control was DNA purified from infected SiHa cells and HeLa cells, respectively.
Immunohistochemisty (IHC)
IHC was used to detect the FHIT protein expression and p53 mutation. The rabbit anti-human FHIT polyclonal antibody and the SP kit were produced by Thermo Fisher Scientific, Inc. Phosphate-buffered saline was used as a negative control instead of the primary antibody. The IHC was undertaken according to the manufacturer's instructions. The results were evaluated independently by three observers. FHIT protein was expressed in the cytoplasm. Staining of FHIT loss was characterized by no cytoplasmic staining in the tumor cells. Each slide was evaluated in 10 visual fields under a microscope (BH-2 Optical Microscope, Olympus Corporation, Tokyo, Japan) at x400 magnification. In each visual field, 100 cells were evaluated, resulting in a total of 1,000 cells per section; the percentage of positive expression of each section was then calculated from these observations. The methods for detecting and evaluating mutations in p53 have been previously described (15).
Statistical analysis
The SPSS software program, version 17.0 (SPSS, Inc., Chicago, IL, USA) was used to apply the χ2 test, Fisher's exact test and Pearson correlation test for statistical analysis. P<0.05 was considered to indicate a statistically significant difference. The demographic characteristics review and statistical analysis were approved by the Department of Public Health, Xi'an Jiaotong University School of Medicine.
Results
Detection of HPVL1/16/18 infection
In the lung cancer cases and non-cancer controls, HPVL1 was detected in 100/180 (55.56%) and 8/110 (7.27%) cases, HPV16 was detected in 67/180 (37.22%) and 7/110 (6.36%) cases and HPV18 was detected in 56/180 (31.11%) and 6/110 (5.45%) cases, respectively. The HPVL1, HPV16 and HPV18 detection rate was increased in the lung cancer group compared with the non-cancer control group (P<0.001, P<0.001 and P<0.001, respectively) (Table II).
Table II.
Detection of HPVL1/16/18 infection.
| Type | Lung cancer cases | Non-cancer controls | P-value |
|---|---|---|---|
| HPVL1+ | 100 (55.56) | 8 (7.27) | <0.001 |
| HPVL1− | 80 (44.44) | 102 (92.73) | |
| HPV16+ | 67 (37.22) | 7 (6.36) | <0.001 |
| HPV16− | 113 (62.78) | 103 (93.64) | |
| HPV18+ | 56 (31.11) | 6 (5.45) | <0.001 |
| HPV18− | 124 (68.89) | 104 (94.55) |
HPV, human papillomavirus.
Association between HPV16 infection, loss of FHIT and pathological characteristics in lung cancer
The HPV16 detection rate was increased in the lung cancer group compared with the non-cancer control group (P<0.001; Table III). HPV16 detection was not correlated with the degree of cell differentiation, tumor-node-metastasis (TNM) stage, lymph node metastasis, gender or age (P>0.05); however, HPV infection was significantly associated with the histological type (P=0.006) and smoking history (P=0.033). The HPV16 detection rate was increased in the cases with lung SCC (51.14%) and those with a smoking history (44.35%) compared with the cases of AC (26.79%), SCLC (19.44%) and those with no smoking history (21.43%). The rate of FHIT loss was increased in the lung cancer group (44.44%) compared with the non-cancer control group (7.27%) (P<0.001). In the lung cancer group, FHIT loss was not correlated with TNM stage, lymph node metastasis, gender or age (P>0.05); however FHIT loss was significantly associated with the histological type (P=0.003), the degree of cell differentiation (P=0.007) and a smoking history (P<0.001). The rate of FHIT loss was increased in the cases of lung SCC (60.23%), and in those with a low degree of cell differentiation (57.65%) or a smoking history (55.65%) compared with the cases of SCLC (33.33%), AC (26.79%) or those with a moderate degree of cell differentiation (38.60%), high degree of cell differentiation (23.68%) or no smoking history (19.64%) (Table III). In addition, the coexistence rate of FHIT loss and a smoking history in the 180 lung cancer cases was 38.33% (69/180) and the Pearson contingency coefficient was r=0.318, (P<0.001) (Table IV).
Table III.
Associations between HPV16 infection, FHIT loss and clinicopathological characteristics in lung cancer.
| Characteristic | HPV16+ | HPV16− | P-value | FHIT loss+ | FHIT loss− | P-value |
|---|---|---|---|---|---|---|
| Non-cancer controls | 7 (6.36) | 103 (93.64) | <0.001 | 8 (7.27) | 102 (92.73) | <0.001 |
| Lung cancer cases | 67 (37.22) | 113 (62.78) | 80 (44.44) | 100 (55.56) | ||
| SCC | 45 (51.14) | 43 (48.86) | 0.006 | 53 (60.23) | 35 (39.77) | 0.003 |
| AC | 15 (26.79) | 41 (73.21) | 15 (26.79) | 41 (73.21) | ||
| SCLC | 7 (19.44) | 29 (80.56) | 12 (33.33) | 24 (66.67) | ||
| Cell differentiation | 0.052 | 0.007 | ||||
| Poor | 39 (45.88) | 46 (54.12) | 49 (57.65) | 36 (42.35) | ||
| Moderate | 21 (36.84) | 36 (63.16) | 22 (38.60) | 35 (61.40) | ||
| Well | 7 (18.42) | 31 (81.58) | 9 (23.68) | 29 (76.32) | ||
| TNM stage of tumors | 0.723 | 0.654 | ||||
| I+II | 43 (38.39) | 69 (61.61) | 45 (40.18) | 67 (59.82) | ||
| III+IV | 24 (35.29) | 44 (64.71) | 35 (51.47) | 33 (48.53) | ||
| Lymph node metastasis | 0.997 | 0.965 | ||||
| N0 | 36 (37.50) | 60 (62.50) | 42 (43.75) | 54 (56.25) | ||
| N1–3 | 31 (36.91) | 53 (63.09) | 38 (45.24) | 46 (54.76) | ||
| Smoking history | 0.033 | <0.001 | ||||
| Yes | 55 (44.35) | 69 (55.65) | 69 (55.65) | 55 (44.35) | ||
| No | 12 (21.43) | 44 (78.57) | 11 (19.64) | 45 (80.36) | ||
| Gender | 0.706 | 0.162 | ||||
| Male | 47 (37.01) | 80 (62.99) | 65 (51.18) | 62 (48.82) | ||
| Female | 20 (37.74) | 33 (62.26) | 15 (28.30) | 38 (71.70) | ||
| Age, years | 0.340 | 0.866 | ||||
| <55 | 32 (38.10) | 52 (61.90) | 39 (46.43) | 45 (53.57) | ||
| ≥55 | 35 (36.46) | 61 (63.54) | 41 (42.71) | 55 (57.29) |
HPV, human papillomavirus; FHIT, fragile histidine triad; SCC, squamous cell carcinoma; AC, adenocarcinoma; SCLC, small cell carcinoma; TNM, tumor-node-metastasis.
Table IV.
Correlation between FHIT loss and smoking history in lung cancer cases.
| Parameter | Smoking+ | Smoking− | Total | χ2 | r |
|---|---|---|---|---|---|
| FHIT loss+ | 69 | 11 | 80 | ||
| FHIT loss− | 55 | 45 | 100 | ||
| Total | 124 | 56 | 180 | 20.251 | 0.318 |
FHIT, fragile histidine triad.
FHIT loss and p53 mutation in lung cancer cases and non-cancer controls
For the lung cancer cases and non-cancer controls, the rate of FHIT loss in the HPVL1-positive group (69.00% and 62.50%, respectively) was significantly increased compared with that in the HPV-negative group (42.50 and 8.82%; P=0.002 and P<0.001, respectively; Table V). In the lung cancer cases, the rate of FHIT loss in the HPV16- and 18-positive groups (77.61 and 71.43%, respectively) was significantly increased compared with the rate in the HPV16- and 18-negative groups (47.79 and 49.19%; P=0.003 and P=0.029, respectively).
Table V.
FHIT loss and p53 mutation in lung cancer cases and non-cancer controls.
| HPV | FHIT loss+ | FHIT loss− | P-value | p53+ | p53− | P-value |
|---|---|---|---|---|---|---|
| Non-cancer controls | ||||||
| HPVL1+ | 5 (62.50) | 3 (37.50) | <0.001 | 1 (12.50) | 7 (87.50) | 0.987 |
| HPVL1− | 9 (8.82) | 93 (91.18) | 11 (10.78) | 91 (89.22) | ||
| Lung cancer cases | ||||||
| HPVL1+ | 69 (69.00) | 31 (31.00) | 0.002 | 71 (71.00) | 29 (29.00) | 0.016 |
| HPVL1− | 34 (42.50) | 46 (57.50) | 39 (48.75) | 41 (51.25) | ||
| HPV16+ | 52 (77.61) | 15 (22.39) | 0.003 | 51 (76.12) | 16 (23.88) | 0.014 |
| HPV16− | 54 (47.79) | 59 (52.21) | 59 (52.21) | 54 (47.79) | ||
| HPV18+ | 40 (71.43) | 16 (28.57) | 0.029 | 35 (62.50) | 21 (37.50) | 0.072 |
| HPV18− | 61 (49.19) | 63 (50.81) | 54 (43.55) | 70 (56.45) |
FHIT, fragile histidine triad; HPV, human papillomavirus.
IHC was used to investigate p53 mutation in the lung tissues. In the non-cancer controls, no significant difference was observed in the p53 mutation rate between the HPVL1-positive group and the HPVL1-negative group (P=0.987). By contrast, in the lung cancer cases, the p53 mutation rate in the HPVL1- and HPV16-positive groups (71.00 and 76.12%, respectively) was significantly increased compared with the HPVL1- and HPV16-negative groups (48.75 and 52.21%; P=0.016 and P=0.014, respectively). However, no significant difference in p53 mutation was observed between the HPV18-positive group and the HPV18-negative group (P=0.072) (Table V).
Correlation between FHIT loss and p53 mutation in HPV-positive lung cancer cases
After obtaining the results showing that HPV infection correlated with FHIT loss and p53 mutation in the lung tissues, it was also observed that FHIT loss coexisted with p53 mutation in 57.00% (57/100) of the HPV-positive lung cancer cases. Due to the high coexistence of FHIT loss and p53 mutation in the HPV-positive lung cancer cases, a correlative study of them was conducted. The analysis demonstrated that FHIT loss and p53 mutation possessed synergistic effects in the lung cancer cases with HPV-infection (Pearson contingency coefficient, r=0.357; P<0.001) (Table VI; Fig. 1).
Table VI.
Correlation between FHIT loss and p53 mutation in HPV-positive lung cancer cases.
| Parameter | p53 mutation+ | p53 mutation− | Total | χ2 | r |
|---|---|---|---|---|---|
| FHIT loss+ | 57 | 12 | 69 | ||
| FHIT loss− | 14 | 17 | 31 | ||
| Total | 71 | 29 | 100 | 14.568 | 0.357 |
FHIT, fragile histidine triad; HPV, human papillomavirus.
Figure 1.
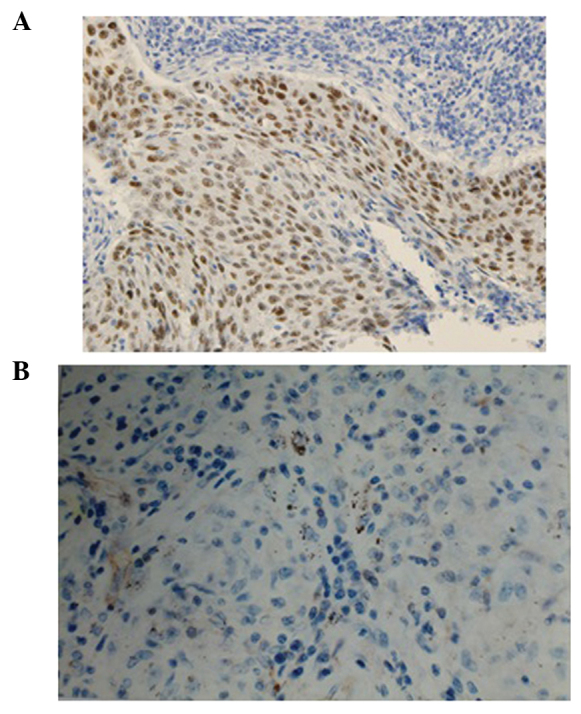
Immunostaining in lung squamous cell carcinoma (magnification, x400). (A) p53 positive expression in lung squamous cell carcinoma. (B) Fragile histidine triad loss in lung squamous cell carcinoma.
Discussion
HPV infection, particularly high-risk HPV16/18 infection, has been identified as a risk factor for the development of cervical cancer, lung cancer and other malignant tumors (1,2,17,18). Previous studies have reported that the involvement of HPV16/18 infection in lung tumorigenesis may be mediated through FHIT and p53 (7,13,14,19,20). However, the interaction between HPV and FHIT is not fully understood, and the association between p53 and FHIT in HPV-infected lung cancer has not been well studied. Therefore the present study aimed to investigate this area of research.
The present study demonstrated that HPV infection was more common in lung cancer cases compared with non-cancer controls (P<0.001). The predominant genotypes were the high-risk HPV16 and/or HPV18 genotypes. HPV16 detection was not correlated with the degree of cell differentiation, TNM stage, lymph node metastasis, gender or age (P>0.05), however, HPV16 infection was significantly associated with the histological type (P=0.006) and smoking history (P=0.033). The HPV16 detection rate was increased in the cases of lung SCC (51.14%) and in those with a smoking history (44.35%) compared with the cases of AC (26.79%), SCLC (19.44%) and those with no smoking history (21.43%). Similar to previous findings (6), the present study indicated that HPV, particularly high-risk HPV16/18, may be responsible for lung cancer development.
The integration of high-risk HPV16/18 DNA genotypes into the host chromosome from the fragile site, FRA3B, which is adjacent to FHIT, is crucial in HPV-induced cervical carcinogenesis (21). As an important tumor suppressor gene, FHIT protein inhibits tumor progression with a wide range of tumor suppressive functions. FHIT loss consequently leads to the development of tumors (10,12,22). Therefore, the present study further investigated FHIT loss in association with HPV infection and the tumor behavior of lung cancer.
The results of the present study demonstrated that the rate of FHIT loss was increased in the lung cancer group compared with the non-cancer control group (P<0.001), indicating that FHIT loss was associated with lung cancer. Pathological analysis demonstrated that FHIT loss was not correlated with the TNM stage of tumors, lymph node metastasis, gender or age (P>0.05); however, FHIT loss was significantly associated with the histological type (P=0.003), degree of cell differentiation (P=0.007) and smoking history (P<0.001). FHIT loss in cases of lung SCC, a low degree of cell differentiation or a smoking history was significantly increased compared with FHIT loss in cases of AC, a high degree of cell differentiation or no smoking history. This finding indicated that FHIT loss may be important in the occurrence of lung cancer, particularly in lung SCC.
Previous studies identified that the FRA3B fragile site, which is covered by FHIT, is targeted by tobacco (23). Tobacco exposure results in an increased level of chromosome fragility at the FRA3B fragile site. The fragility of the chromosome may be related to abnormalities in replication, resulting in single-strand DNA gaps that, if unrepaired, may lead to chromosomal damage, including the deletion of distal genes, deletions within the fragile site, and translocations or other rearrangements with a break at the FRA3B fragile site. FHIT gene loss or a reduction in its expression may be associated with the instability of FRA3B (24). Therefore, this may explain the observation that the rate of FHIT loss was increased in the cases with a history of smoking compared with no smoking history in the present study.
A previous study of cervical cancer demonstrated that FHIT anomalies were closely associated with high-risk HPV16/18 infection (23,24). The results of the present study demonstrated that FHIT loss was also associated with HPV infection in lung cancer. FHIT loss in the HPV-positive group was significantly increased compared with the HPV-negative group. It was similar in the HPV16/18-positive group and the HPV16/18-negative group. These results demonstrated that FHIT loss is associated with HPV infection, particularly high-risk HPV16/18 infection. The present study further indicated the role of FHIT in the tumorigenesis process and may also assist in the understanding of the role of FHIT in lung cancer. In vitro studies have demonstrated that HPV is able to insert its genes into the fragile site FRA3B adjacent to FHIT resulting in allele loss of the gene (12). Therefore, it may reasonably be considered that HPV infection, particularly high-risk HPV16/18 infection, may induce a fissure or breaking point in the FRA3B fragile site, into which HPV-DNA may integrate into the host cell (6), resulting in the loss of FHIT protein expression. These observations indicate that FHIT loss may provide the integration site for HPV, while at the same time, the integration of HPV results in the inactivation of FHIT. Therefore, the involvement of HPV infection in lung tumorigenesis may, at least in part, be mediated through FHIT loss.
In the present study, in the non-cancer controls and lung cancer cases, the rate of FHIT loss in the HPV-positive group was significantly increased compared with the HPV-negative group (P<0.001 and P=0.002, respectively). For the lung cancer cases, the rate of FHIT loss in the HPV16- and 18-positive groups was significantly increased compared with the HPV16/18-negative group (P=0.003 and P=0.029, respectively). The coexistence of FHIT loss and a smoking history in the 180 lung cancer cases was 38.33% (69/180) and the Pearson contingency coefficient was r=0.318 (P<0.001). These results may further explain the contributions to the lung tumorigenesis of FHIT loss and indicate that FHIT loss may be an early indicator for lung cancer, particularly for patients with a history of smoking.
Meanwhile, FHIT loss and p53 mutation have previously been observed to frequently occur in lung cancer and may therefore contribute to the development of lung cancer. The present study further investigated p53 mutations using IHC. In the lung cancer cases, the p53 mutation rate in the HPVL1- and HPV16-positive groups was demonstrated to be significantly increased compared with the HPVL1- and HPV16-negative groups (P=0.016 and P=0.014, respectively). These findings indicated that p53 mutation is associated with HPV infection, particularly HPV16 infection. The co-expression studies demonstrated that FHIT loss and p53 mutation had synergistic effects in the HPV-infected lung cancer cases, with a Pearson contingency coefficient of r=0.357 (P<0.001), indicating that FHIT loss and p53 mutation may coordinate in the development of HPV-infected lung cancer.
In light of the findings of the present study and our previous studies (6,14), one hypothesis is that following infection of the lung tissue with HPV, HPV-DNA integrates into the host-cell DNA at the FRA3B fragile site and results in FHIT loss. This integration may result in the upregulation of the transcription of the E6 and E7 oncogenes. E6 expression accelerates p53 mutation (25), which increases the possibility of a HPV-infected cells becoming malignant. Abnormal loss of FHIT occurs early and frequently in the progression from normal to malignant lesions. The results of the present study provide early epidemiological data that require further study in high-risk HPV16/18-infected lung SCC patients.
In summary, the results of the present study indicated that the involvement of HPV infection in lung tumorigenesis may, at least in part, be mediated through FHIT loss. FHIT loss and p53 mutation may be coordinated in the development of HPV-associated lung cancer, and may accelerate the occurrence and development of the disease.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant no. 81273148) and the International Cooperation Foundation of Shaanxi Province (grant no. 2013KW32-05). The authors would like to acknowledge the contributions of Professor Suminori Akiba (Kagoshima University, Japan), who provided assistance in analyzing the data. The authors would also like to thank Dr Guanjun Zhang (Director of the Department of Pathology, the First Affiliated Hospital of Medical School of Xi'an Jiaotong University) for contributing to the pathological diagnosis and for assistance in the preparation of the paraffin-embedded slices.
References
- 1.Al-Badawi IA, Al-Suwaine A, Al-Aker M, Asaad L, Alaidan A, Tulbah A, Fe Bohol M, Munkarah AR. Detection and genotyping of human papilloma virus in cervical cancer specimens from Saudi patients. Int J Gynecol Cancer. 2011;21:907–910. doi: 10.1097/IGC.0b013e318214219f. [DOI] [PubMed] [Google Scholar]
- 2.Al-Shabanah OA, Hafez MM, Hassan ZK, Sayed-Ahmed MM, Abozeed WN, Al-Rejaie SS, Alsheikh AA. Human papillomavirus genotyping and integration in ovarian cancer Saudi patients. Virol J. 2013;10:343–352. doi: 10.1186/1743-422X-10-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang X, Lu L. Expression of HPV-16 E6 protein and p53 inactivation increases the uterine cervical cancer invasion. Drug Res (Stuttg) 2015;65:70–73. doi: 10.1055/s-0034-1372614. [DOI] [PubMed] [Google Scholar]
- 4.Aumayr K, Susani M, Horvat R, Wrba F, Mazal P, Klatte T, Koller A, Neudert B, Haitel A. P16INK4A immunohistochemistry for detection of human papilloma virus-associated penile squamous cell carcinoma is superior to in-situ hybridization. Int J Immunopathol Pharmacol. 2013;26:611–620. doi: 10.1177/039463201302600305. [DOI] [PubMed] [Google Scholar]
- 5.Sarchianaki E, Derdas SP, Ntaoukakis M, et al. Detection and genotype analysis of human papillomavirus in non-small cell lung cancer patients. Tumour Biol. 2014;35:3203–3209. doi: 10.1007/s13277-013-1419-2. [DOI] [PubMed] [Google Scholar]
- 6.Yu Y, Yang A, Hu S, Yan H. Correlation of HPV-16/18 infection of human papillomavirus with lung squamous cell carcinomas in Western China. Oncol Rep. 2009;21:1627–1632. doi: 10.3892/or_00000397. [DOI] [PubMed] [Google Scholar]
- 7.Jafari H, Gharemohammadlou R, Fakhrjou A, et al. Genotyping of human papillomavirus and TP53 mutations at exons 5 to 7 in lung cancer patients from Iran. Bioimpacts. 2013;3:135–140. doi: 10.5681/bi.2013.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ragin C, Obikoya-Malomo M, Kim S, et al. HPV-associated lung cancers: An international pooled analysis. Carcinogenesis. 2014;35:1267–1275. doi: 10.1093/carcin/bgu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasegawa Y, Ando M, Kubo A, Isa S, Yamamoto S, Tsujino K, Kurata T, Ou SH, Takada M, Kawaguchi T. Human papilloma virus in non-small cell lung cancer in never smokers: A systematic review of the literature. Lung Cancer. 2014;83:8–13. doi: 10.1016/j.lungcan.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Ohta M, Inoue H, Cotticelli MG, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84:587–597. doi: 10.1016/S0092-8674(00)81034-X. [DOI] [PubMed] [Google Scholar]
- 11.Wistuba II, Montellano FD, Milchgrub S, et al. Deletions of chromosome 3p are frequent and early events in the pathogenesis of uterine cervical carcinoma. Cancer Res. 1997;57:3154–3158. [PubMed] [Google Scholar]
- 12.Wilke CM, Hall BK, Hoge A, Paradee W, Smith DI, Glover TW. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: Direct evidence for the coincidence of viral integration sites and fragile sites. Hum Mol Genet. 1996;5:187–195. doi: 10.1093/hmg/5.2.187. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Cheng YW, Wu DW, Chen JT, Chen CY, Chou MC, Lee H. Frequent FHIT gene loss of heterozygosity in human papillomavirus-infected non-smoking female lung cancer in Taiwan. Cancer Lett. 2006;235:18–25. doi: 10.1016/j.canlet.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 14.Yu Y, Yang A, Hu S, Zhang J, Yan H. Significance of human papillomavirus 16/18 infection in association with p53 mutation in lung carcinomas. Clin Respir J. 2013;7:27–33. doi: 10.1111/j.1752-699X.2011.00277.x. [DOI] [PubMed] [Google Scholar]
- 15.Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greer CE, Wheeler CM, Manos MM. PCR amplification from paraffin-embedded tissues: sample preparation and the effects of fixation. In: Carl WD, Gabriela SD, editors. PCR Primer: a laboratory manual. Cold Spring Harbor Laboratory Press; New York: 1995. pp. 99–112. [Google Scholar]
- 17.Wang JL, Fang CL, Wang M, Yu MC, Bai KJ, Lu PC, Liu HE. Human papillomavirus infections as a marker to predict overall survival in lung adenocarcinoma. Int J Cancer. 2014;134:65–71. doi: 10.1002/ijc.28349. [DOI] [PubMed] [Google Scholar]
- 18.Badillo-Almaraz I, Zapata-Benavides P, Saavedra-Alonso S, Zamora-Avila D, Reséndez-Pérez D, Tamez-Guerra R, Herrera-Esparza R, Rodríguez-Padilla C. Human papillomavirus 16/18 infections in lung cancer patients in Mexico. Intervirology. 2013;56:310–315. doi: 10.1159/000351075. [DOI] [PubMed] [Google Scholar]
- 19.Iwakawa R, Kohno T, Enari M, Kiyono T, Yokota J. Prevalence of human papillomavirus 16/18/33 infection and p53 mutation in lung adenocarcinoma. Cancer Sci. 2010;101:1891–1896. doi: 10.1111/j.1349-7006.2010.01622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bahnassy AA, Zekri AR, Madbouly MS, El-Naggar M, El-Khelany ZF, El-Merzebany MM. The correlation between FHIT, P53 and MMR genes in human papillomavirus-associated cervical carcinoma. J Egypt Natl Canc Inst. 2006;18:191–202. [PubMed] [Google Scholar]
- 21.Giarnieri E, Zanesi N, Bottoni A, Alderisio M, Lukic A, Vecchione A, Ziparo V, Croce CM, Mancini R. Oncosuppressor proteins of fragile sites are reduced in cervical cancer. Cancer Lett. 2010;289:40–45. doi: 10.1016/j.canlet.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sozzi G, Veronese ML, Neghni M, et al. The FHIT gene 3p14.2 is abnormal in lung cancer. Cell. 1996;85:17–26. doi: 10.1016/S0092-8674(00)81078-8. [DOI] [PubMed] [Google Scholar]
- 23.Kapitanović S, Čačev T, Lončar B, Catela Ivković T, Križanac Š, Pavelić K. Reduced FHIT expression is associated with tumor progression in sporadic colon adenocarcinoma. Exp Mol Pathol. 2014;96:92–97. doi: 10.1016/j.yexmp.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Stein CK, Glover TW, Palmer JL, Glisson BS. Direct correlation between FRA3B expression and cigarette smoking. Genes Chromosomes Cancer. 2002;34:333–340. doi: 10.1002/gcc.10061. [DOI] [PubMed] [Google Scholar]
- 25.Neyaz MK, Hussain S, Hassan MI, et al. Novel missense mutation in FHIT gene: Interpreting the effect in HPV-mediated cervical cancer in Indian women. Mol Cell Biochem. 2010;335:53–58. doi: 10.1007/s11010-009-0240-0. [DOI] [PubMed] [Google Scholar]