

Abstract
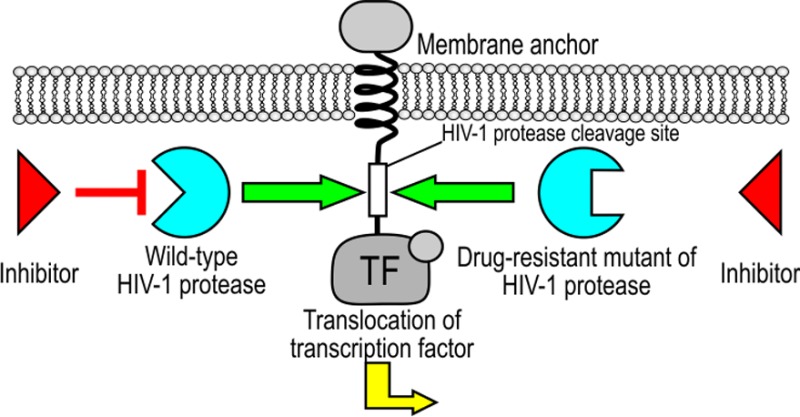
The high mutation rate of the human immunodeficiency virus type 1 (HIV-1) virus is a major problem since it evades the function of antibodies and chemical inhibitors. Here, we demonstrate a viral detection strategy based on synthetic biology principles to detect a specific viral function rather than a particular viral protein. The resistance caused by mutations can be circumvented since the mutations that cause the loss of function also incapacitate the virus. Many pathogens encode proteases that are essential for their replication and that have a defined substrate specificity. A genetically encoded sensor composed of a fused membrane anchor, viral protease target site, and an orthogonal transcriptional activator was engineered into a human cell line. The HIV-1 protease released the transcriptional activator from the membrane, thereby inducing transcription of the selected genes. The device was still strongly activated by clinically relevant protease mutants that are resistant to protease inhibitors. In the future, a similar principle could be applied to detect also other pathogens and functions.
Keywords: synthetic biology, genetic device, HIV-1 protease, protease-activated transcription factor, drug-resistant mutants
Viral diseases represent a significant challenge for medical therapy, and the human immunodeficiency virus type 1 (HIV-1) is one of the most difficult viruses to treat because it hijacks the cells of the immune system. HIV-1 uses the viral glycoproteins gp120 and gp41 to bind to cellular receptors, reverse transcriptase to transcribe its RNA into DNA, integrase to integrate viral genome into the host, and viral protease to cleave viral polyprotein into functional proteins. All of these viral proteins have been and still are investigated as targets for therapeutic inhibitors to interfere with the viral lifecycle. The main reason for the difficulty in developing effective drugs is the lack of proofreading in the reverse transcriptase of HIV-1. As a consequence, mutations occur with high frequency, thereby modifying the sequence of the HIV-1 proteins. Resistance evolves quickly, rendering many of the chemical inhibitors or antibodies useless. The most effective current treatment is based on highly active antiretroviral therapy, which employs a cocktail of several antiretroviral drugs against different targets based on different inhibition mechanisms, thereby decreasing the probability of simultaneous mutational escape.1
HIV-1 viral proteins are translated as a Gag-Pol-polyprotein that is cleaved by the viral protease into 11 polypeptide chains. Without this proteolytic cleavage, the virus remains noninfectious.2 Dimeric aspartic viral protease, which cleaves this polyprotein, is specific to the virus, and proteases from human cells cannot supplement its function.3 Random mutations of HIV-1 protease that abolish its activity—or the pharmacological inhibition—disrupt HIV’s ability to replicate and infect additional cells.4 HIV-1 protease was one of the first targets of therapy, and protease inhibitors are still being used in treatment. However, mutations that occur within the protease may retain a sufficient fraction of the proteolytic activity that supports viral survival and renders the protease resistant to inhibitors. The rapid mutation rate of the HIV-1 virus results in a large number of variants, even within an individual patient, and the selective pressure of antiviral therapy leads to the proliferation of drug-resistant HIV-1 protease strains. The G48V mutant is considered to be the key signature residue mutation of HIV-1 protease in the development of resistance against saquinavir (SAQ) therapy.5 G48V usually co-occurs with the resistance mutations I54T and V82A.6 Mutations at position 54 contribute to the resistance of the virus against all of the approved protease inhibitors,7 and the V82A mutant is one of the first drug-resistant mutations to occur in HIV-1 protease in patients receiving antiviral therapy.8,9
In this study, we have considered the possibility of employing synthetic biology principles to engineer a cellular device that can detect the HIV-1 infection and trigger production of reporter or antiviral proteins. We reasoned that an effective biological antiviral device should be based on essential viral function rather than on a specific protein with a defined sequence or tertiary structure that is targeted by chemical inhibitors or antibodies. By relying on the viral function, the viral detection should be less sensitive to mutations. One possible use of this device could be for the production of effectors that can alert the human immune system for the presence of a virus (e.g., the production of interferons [IFNs], chemokines). Once the viral activity is detected, the device should be able to transduce this signal to activate the selected effector. The signaling device should produce a robust and specific signal.
The principle behind the genetically encoded device for the detection of the viral protease activity is based on the proteolytic cleavage of the peptide linker, which links the orthogonal transcriptional activator Gal4-VP16 to the membrane anchor. The viral presence (specifically the viral protease activity) is sensed by the irreversible proteolytic step, which causes the release of the transcriptional activator Gal4-VP16 from the membrane and activation of the selected transcriptional response. The Gal4-VP16 activator, therefore, exists in two states: an inactive state, where the transcriptional activator is located at the membrane and where it cannot bind to the respective operator site, and an active state, where the Gal4-VP16 is translocated into the nucleus where it can bind to and activate the selected effector gene (e.g., reporter or human IFN-β1).
We engineered a hybrid transcription factor (Gal4-VP16, which is composed of the DNA-binding domain [Gal4] and the activation domain [VP16]) to be linked through the HIV-1 protease cleavage site to the cellular membrane by the transmembrane and ectodomains of the CD4 or FAS receptors (Figure 1). Yeast regulatory protein Gal4,10 which is comprised of a DNA-binding domain, linker, and a coiled-coil dimerization domain, has been used previously as an orthogonal transcription factor in mammalian cells.11 A domain of the herpes simplex virus protein VP16 recruits host-encoded elements of the transcriptional machinery upstream from the promoter.12,13 The selected peptide linker was comprised of the HIV-1 protease cleavage site with the amino acid sequence SQVSQNY↓PIVQNLQ, which is also called the p17/p24 peptide14 and is used as the synthetic HIV Protease Substrate 1.15 For an efficient anchor to the membrane, we selected the nonfunctional transmembrane and ectodomains of CD4 and FAS, which are type-I transmembrane receptors that are found on the surface of immune cells, such as T-helper cells, monocytes, macrophages, and dendritic cells.16,17
Figure 1.

Design of a mutation-resistant antiviral signaling device based on HIV-1 protease activity. The transcriptional activator Gal4-VP16 is anchored to the transmembrane protein domain (CD4 or FAS), where it is inactive. The activation step occurs via the HIV-1 protease, which cleaves the linker between the transcription factor and membrane anchor, thereby comprising the HIV-1 protease cleavage site. After the irreversible proteolytic step, Gal4-VP16 is released from the membrane and translocates into the nucleus, where it transcribes the selected genes under the control of the GAL4 operator, such as the reporter genes or genes that provide defense against viral infections.
Activation of the device by the viral protease was observed with fluorescence confocal microscopy, where we used the fluorescent proteins cyan florescent protein (CFP) or mCherry linked by a cleavable linker peptide (a HIV-1 protease cleavage site [hivp]) to the cytosolic side of the transmembrane domain. Human embryonic kidney (HEK)293T cells were transfected with the plasmid coding fusion proteins CD4(HA)-hivp-CFP-6xHis or FAS(HA)-hivp-mCherry-2xNLS and cotransfected with a replication-incompetent HIV-1 pseudoviral genome on a pNL4–3.HSA.R–.E– plasmid, thereby encoding the functional viral protease. In the absence of the HIV-1 genome, the fluorescence was clearly localized to the plasma membrane (Figure 2a and c). In the cells cotransfected with an HIV-1 genome, the fluorescent CFP was released from the membrane into the cytosol (Figure 2b), while mCherry, which is comprised of two copies of SV40 large T-antigen nuclear localization sequence (NLS), was translocated to the nucleus (Figure 2d). The expression of the transmembrane proteins CD4(HA)-hivp-GAL4-VP16 or FAS(HA)-hivp-GAL4-VP16 at the surface of the cell membrane was also confirmed by staining the HEK293T cells with anti-HA antibodies, as the HA-tag was positioned at the extracellular side of the receptors (Supporting Information Figure S1). The incorporation of the transmembrane receptors into the membrane demonstrates the appropriate protein folding and localization, suggesting that the sensor could be functional.
Figure 2.

HIV-1 protease triggers the release of the membrane-bound reporter. In cells without HIV-1 protease, the membrane localization of (a) CFP and (c) mCherry-2xNLS was observed. In the presence of HIV-1 protease, (b) CFP was released to the cytosol and (d) mCherry was released from the membrane and translocated to the nucleus. An overlay of the image showing the nucleus stained with Hoechst 33342 and mCherry is shown on the left (d). The microscopic images are representative of five separate observations. Scale bar = 10 μm. The HEK293T cells were transfected with (a, b) 40 ng CD4(HA)-hivp-CFP-6xHis or (c, d) 40 ng CD4(HA)-hivp-mCherry-2xNLS, as well as with the (b, d) HIV-1 genome-encoding plasmid pNL4–3.HSA.R–.E– (320 or 360 ng).
We demonstrated the sensing and transcriptional activation of the device using a luciferase reporter assay, with which we measured the expression of the luciferase gene induced by the transcriptional activator, Gal4-VP16. The transcriptional activity of Gal4-VP16 was triggered by the cleavage of the linker to the transmembrane receptor by the HIV-1 protease, which released Gal4-VP16 into the nucleus and activated the transcription of the luciferase under the control of the Gal4-responsive operator. The luciferase activity was observed only in cells cotransfected with the HIV-1 genome, and the plasmid for CD4(HA)-hivp-GAL4-VP16 (Supporting Information Figure S2a) or FAS(HA)-hivp-GAL4-VP16 (Supporting Information Figure S2b). Synthetic HIV-1 protease inhibitors inhibited the activation of Gal4-VP16, as in the presence of 5 μM SAQ or 5 μM ritonavir (RTV), activation of the reporter was reduced by more than 70% for CD4(HA)-hivp-GAL4-VP16 (Supporting Information Figure S2a) and more than 90% for FAS(HA)-hivp-GAL4-VP16 (Supporting Information Figure S2b).
The salient feature of our device is that it should be less sensitive to the mutations that cause resistance to protease inhibitors. The viral genome encoding drug-resistant HIV-1 protease mutants G48V/I54T and V82A exhibited slightly reduced proteolytic activity in comparison to the wild type for both device constructs, CD4(HA)-hivp-GAL4-VP16 or FAS(HA)-hivp-GAL4-VP16 (Supporting Information Figure S3), probably because of the impaired specific activity of the mutated protease. However, their proteolytic efficiency is still sufficient for processing the viral polyprotein and viral replication. Both clinically observed HIV-1 protease mutants were significantly less sensitive to the therapeutic protease inhibitors or their cocktails (atazanavir, SAQ, and RTV) than the wild-type HIV-1 protease (Figure 3d–f). However, our device was able to robustly sense the activity of both HIV-1 protease mutants and trigger gene transcription regardless of the presence of 1 μM SAQ or a cocktail of 1 μM SAQ and 1 μM RTV, as shown in Figure 3a–c for the CD4(HA)-hivp-GAL4-VP16 device. We obtained similar results for FAS(HA)-hivp-GAL4-VP16 (these data are not shown). These findings demonstrate that the synthetic genetic device is still activated by the protease mutations that are resistant to clinically used inhibitors. We have also demonstrated the activation of our device with nontoxic HIV-1 protease precursor fused to green fluorescent protein (GFP).18 For the activation of the device in HEK293T cells, significantly lower amounts of GFP-PR in comparison to the pNL4–3.HSA.R–.E– were required (Supporting Information Figure S4). The protease mutant GFP-PR(G48V/I54T) was also sensed by our device. We found that mutant GFP-PR(G48V/I54T) was almost completely resistant to 1 μM SAQ, and we used Western blot to demonstrate the expression levels of both precursor proteins (Supporting Information Figure S5). To demonstrate that our device could drive expression of different selected effector genes, we selected the gene for human IFN-β1. IFN-β1 is a member of the type I IFN family; these IFNs are usually secreted as the consequence of elevated levels of dsRNA in cells and are used in antiviral therapy.19 We found that the expression level of IFN-β1 that was produced in HEK293T cells—transfected with our signaling device—was dependent on the amount of transfected HIV-1 protease (Figure 4).
Figure 3.

Synthetic anti-HIV-1 signaling device detects the activity of HIV-1 protease mutations resistant to protease inhibitors. (a, b, c) HEK293T cells were transfected with a reporter plasmid coding for the firefly luciferase, a plasmid constitutively expressing the Renilla luciferase, 10 ng of CD4(HA)-hivp-GAL4-VP16, and increasing amounts of pNL4–3.HSA.R–.E– (pNL4–3 in the figure) or HIV-1 protease mutants (G48V/I54T or V82A) in the medium with or without saquinavir (SAQ) or saquinavir and ritonavir (SAQ/RTV). The cells were lysed 24 h after transfection, and the expression of the firefly and Renilla luciferase reporter genes was analyzed. (d, e, f) HEK293T cells were transfected with plasmids expressing the firefly luciferase reporter and the constitutively expressed Renilla luciferase, CD4(HA)-hivp-GAL4-VP16 (10 ng) and pNL4–3.HSA.R–.E–, G48V/I54T, or V82A (all 120 ng) in the medium with or without the HIV-1 protease inhibitors atazanavir (ATZ), atazanavir and ritonavir (ATZ/RTV), saquinavir (SAQ), or saquinavir and ritonavir (SAQ/RTV). The cells were lysed 24 h after transfection, and the activity of the firefly and Renilla luciferase reporters was measured. Error bars represent the SD obtained from four experimental replicates. The data are representative of two or three independent experiments. The two-tailed Student’s t-test (assuming a two-sample equal variance) was used for pairwise comparisons. ***p < 0.005.
Figure 4.
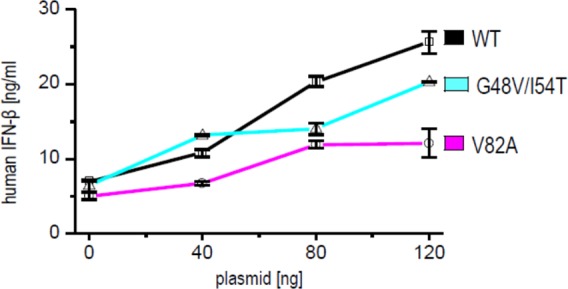
Synthetic anti-HIV-1 signaling device triggers production of human IFN-β1 in response to the viral protease. HEK293T cells were transfected with an effector plasmid pABM2(IFN-β1) coding for human IFN-β1, 10 ng of CD4(HA)-hivp-GAL4-VP16, and increasing amounts of pNL4–3.HSA.R–.E– (WT in the figure) or HIV-1 protease mutants (G48V/I54T or V82A). IFN-β concentrations (ng/mL ± SD) were determined through a comparison with a human IFN-β standard curve. The data are representative of three independent experiments with duplicate samples.
Our work presents an orthogonal cellular signaling device that is activated by the specific protease from HIV-1. The use of the HIV-1 protease to trigger the selected activity has been previously reported4 but not for triggering the cellular genetic response program. In addition, eukaryotic signaling pathways have been previously rewired using synthetic biology.20 Daringer et al. recently reported a synthetic device in mammalian cells that senses extracellular ligands through ligand binding-induced dimerization, which releases an orthogonal transcription factor that is independent of native cellular components.21 Engineering sensing devices may also serve as tools to address the difficult problem of the host defense against rapidly mutating viruses or viruses that avoid immune surveillance. The detection of specific viral functions by coupling to a synthetic signaling pathway could be utilized to trigger almost any selected transcriptional response. For antiviral defense using our device, the reporter gene is replaced with one or several effector genes. We have demonstrated that our system is able to trigger the production of human IFN-β1, which can serve as an “alarm” that signals neighboring cells to defend against a viral infection. The amount of produced IFN-β1 can reach up to 25 ng/mL, which is several orders of magnitude higher than the systemic IFN concentration in antiviral therapy (e.g., 0.2 mg/day every other day for the treatment of hepatitis C virus [HCV]).22 One potential application of our synthetic device could be as a detection assay for screening the library of drug-resistant HIV-1 protease mutants and inhibitors in mammalian culture cells using different reporters, including cell toxic proteins as the selection markers. The advantages of using cell line–based assays for HIV-1 protease activity have already been reported; authors used different HIV-1 protease precursors (GFP-PR18 or Gal4-PR23) or probe proteins, such as Förster resonance energy transfer-based HIV-1 protease-sensitive sensors.24,25 Through experimental implementation of our signaling device, we have found that it is also faster than two-round phenotyping assays for testing HIV-1 isolates.26
The primary purpose of the described device is the targeting of essential viral function, regardless of the specific sequence or spatial structure of the particular viral components. This approach has an important advantage in comparison to the detection or therapy that targets specific viral proteins through antibodies, chemical inhibitors, or specific interfering RNA molecules. Their efficiency and robustness depends on the selection of an invariant segment that is independent of viral mutations or relies on the statistical advantage based on targeting multiple viral proteins. The described principle is highly specific and could be tailored for any specific protease. Replacing Gal4 with a designable DNA-binding domain (e.g., TAL effector–based activators27,28) could also rewire the signal to regulate the production of selected endogenous genes. The same principle for the function-based mutation-independent signaling system could also be applied to the detection of other pathogens, particularly viruses (e.g., severe acute respiratory syndrome [SARS]; West Nile virus; hand, foot and mouth disease; etc.). Many viral proteases have a well-defined substrate specificity, which could be implemented in a straightforward manner for the design of a linker peptide. For example, HCV protease, a NS3/4A serine protease that is essential for viral replication as it is responsible for proteolytic cleavage of the HCV polyprotein precursor, cleaves specific amino acid sequences.29 The core sequence of the cleavage site for the SARS-coronavirus 3C-like cysteine protease, which is responsible for the maturation of functional viral proteins, is also highly conserved.30 The Murray Valley encephalitis and West Nile virus NS2B/NS3 proteases also have an unusual sequence recognition specificity that is not found in mammalian proteases.31 Finally, the same function-based principle could be used to detect other specific viral functions, such as reverse transcription or integration.
Methods
A detailed description of the methods used in this study can be found under Supporting Information.
DNA Constructs
We prepared the following plasmids: CD4(HA)-hivp-GAL4-VP16, FAS(HA)-hivp-GAL4-VP16, CD4(HA)-hivp-CFP-6xHis, and FAS(HA)-hivp-mCherry-2xNLS (Tables S1–S4). We prepared the HIV-1 protease mutants pNL4–3.HSA.R–.E–(G48V/I54T) (hereafter referred to as G48V/I54T) with the G48V and I54T mutations within the HIV-1 protease sequence and pNL4–3.HSA.R–.E–(V82A) (hereafter referred to as V82A), in which the HIV-1 protease sequence contains a mutation for V82A. The same mutations were also introduced into the vector pcDNA3/GFP-PR (hereafter referred to as GFP-PR) to prepare the construct GFP-PR(G48V/I54T). The effector plasmid pABM2(IFN-β1) was prepared from the plasmid pABM2 with GAL4-binding sites and coding for firefly luciferase, a kind gift from Dr. Mark S. Ptashne.
Luciferase Reporter Assay
The HEK293T cells were harvested from an actively growing culture and plated on 96-well tissue culture plates at 2 × 104 cells per well. After 24 h at approximately 70% confluence, the cells were transfected with appropriate plasmid combinations: a plasmid for fusion protein CD4(HA)-hivp-GAL4-VP16 or FAS(HA)-hivp-GAL4-VP16, a plasmid coding for the (wild type or mutant) HIV-1 protease, reporter plasmid pABM2 (15 ng), and pHRL-TK coding for the constitutive Renilla luciferase transfection control. The cells were harvested 24 or 48 h after transfection. The activities of the firefly luciferase and Renilla luciferase were measured using the dual-luciferase assay.
Confocal Microscopy
HEK293T cells (5 × 104 cells/well) were seeded in 8-well tissue culture chambers. At 30% confluency, the cells were transfected with plasmids: 40 ng of CD4(HA)-hivp-CFP-6xHis or FAS(HA)-hivp-mCherry-2xNLS with or without 320 ng or 360 ng pNL4–3.HSA.R–.E–. The images were acquired 2 days after transfection.
Quantification of IFN-β1 Levels
The HEK293T cells were harvested from an actively growing culture and plated on 96-well tissue culture plates at 2 × 104 cells per well. After 24 h at approximately 70% confluence, the cells were transfected with a plasmid CD4(HA)-hivp-GAL4-VP16 (10 ng), a plasmid coding for (wild type or mutant) HIV-1 protease, and an effector plasmid pABM2(IFN-β1). At 48 or 72 h after transfection, we collected the culture medium and determined the IFN-β1 level in the medium.
Acknowledgments
This research was supported by the Slovenian Research Agency and the EN-FIST Centre of Excellence. We thank the members of the Slovenian iGEM 2007 competition team and Saša Jereb in particular (currently at Rockefeller University, U.S.A.) for their initial work for this study. The following reagents were obtained through the NIH AIDS Reagent Program (Division of AIDS, NIAID): pNL4−3.HSA.R–.E– from Dr. Nathaniel Landau and the HIV-1 protease inhibitors saquinavir, ritonavir, and atazanavir. We thank Dr. Mark S. Ptashne from the Memorial Sloan-Kettering Cancer Center, U.S.A., for the pSGVP and pABM2 plasmids.
Supporting Information Available
Detailed description of the methods, additional microscopic images and charts, as well as tables of nucleotide and amino acid sequences. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
A.M., R.G., and R.J. designed the study; A.M., R.G., and M.B. performed and analyzed the experiments; A.M. and R.J. wrote the manuscript; M.B. assisted with preparing the figures.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hammer S. M.; Eron J. J.; Reiss P.; Schooley R. T.; Thompson M. A.; Walmsley S.; Cahn P.; Fischl M. A.; Gatell J. M.; Hirsch M. S.; Jacobsen D. M.; Montaner J. S.; Richman D. D.; Yeni P. G.; Volberding P. A.; (2008) Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society-U.S.A. panel. JAMA, J. Am. Med. Assoc. 300, 555–570. [DOI] [PubMed] [Google Scholar]
- Kohl N. E.; Emini E. A.; Schleif W. A.; Davis L. J.; Heimbach J. C.; Dixon R. A.; Scolnick E. M.; Sigal I. S. (1988) Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U.S.A. 85, 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brik A.; Wong C. H. (2003) HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 1, 5–14. [DOI] [PubMed] [Google Scholar]
- McPhee F.; Good A. C.; Kuntz I. D.; Craik C. S. (1996) Engineering human immunodeficiency virus 1 protease heterodimers as macromolecular inhibitors of viral maturation. Proc. Natl. Acad. Sci. U.S.A. 93, 11477–11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle J.; Bechowsky B.; Rose D.; Hauser U.; von der Helm K.; Gurtler L.; Nitschko H. (1995) Resistance of HIV type 1 to proteinase inhibitor Ro 31–8959. AIDS Res. Hum. Retroviruses 11, 671–676. [DOI] [PubMed] [Google Scholar]
- Palmer S.; Shafer R. W.; Merigan T. C. (1999) Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS 13, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla A.; Korneyeva M.; Gao Q.; Vasavanonda S.; Schipper P. J.; Mo H. M.; Markowitz M.; Chernyavskiy T.; Niu P.; Lyons N.; Hsu A.; Granneman G. R.; Ho D. D.; Boucher C. A.; Leonard J. M.; Norbeck D. W.; Kempf D. J. (1996) Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat. Med. 2, 760–766. [DOI] [PubMed] [Google Scholar]
- Shafer R. W.; Stevenson D.; Chan B. (1999) Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 27, 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M.; Nalivaika E. A.; King N. M.; Schiffer C. A. (2004) Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 78, 12446–12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giniger E.; Varnum S. M.; Ptashne M. (1985) Specific DNA binding of GAL4, a positive regulatory protein of yeast. Cell 40, 767–774. [DOI] [PubMed] [Google Scholar]
- Elliott D. A.; Brand A. H. (2008) The GAL4 system: A versatile system for the expression of genes. Methods Mol. Biol. 420, 79–95. [DOI] [PubMed] [Google Scholar]
- Wu T. J.; Monokian G.; Mark D. F.; Wobbe C. R. (1994) Transcriptional activation by herpes simplex virus type 1 VP16 in vitro and its inhibition by oligopeptides. Mol. Cell. Biol. 14, 3484–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I.; Ma J.; Triezenberg S.; Ptashne M. (1988) GAL4-VP16 is an unusually potent transcriptional activator. Nature 335, 563–564. [DOI] [PubMed] [Google Scholar]
- Cote H. C.; Brumme Z. L.; Harrigan P. R. (2001) Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J. Virol. 75, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matayoshi E. D.; Wang G. T.; Krafft G. A.; Erickson J. (1990) Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science 247, 954–958. [DOI] [PubMed] [Google Scholar]
- Isobe M.; Huebner K.; Maddon P. J.; Littman D. R.; Axel R.; Croce C. M. (1986) The gene encoding the T-cell surface protein T4 is located on human chromosome 12. Proc. Natl. Acad. Sci. U.S.A. 83, 4399–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant H. (2002) The Fas signaling pathway: More than a paradigm. Science 296, 1635–1636. [DOI] [PubMed] [Google Scholar]
- Lindsten K.; Uhlikova T.; Konvalinka J.; Masucci M. G.; Dantuma N. P. (2001) Cell-based fluorescence assay for human immunodeficiency virus type 1 protease activity. Antimicrob. Agents Chemother. 45, 2616–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel C. E. (2001) Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya R. P.; Remenyi A.; Yeh B. J.; Lim W. A. (2006) Domains, motifs, and scaffolds: The role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75, 655–680. [DOI] [PubMed] [Google Scholar]
- Daringer D.; N. M; Dudek R. M.; Schwarz K. A.; Leonard J. N. (2014) Modular extracellular sensor architecture for engineering mammalian cell-based devices. ACS Synth. Biol. 10.1021/sb400128g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried M. W.; Shiffman M. L.; Reddy K. R.; Smith C.; Marinos G.; Goncales F. L. Jr.; Haussinger D.; Diago M.; Carosi G.; Dhumeaux D.; Craxi A.; Lin A.; Hoffman J.; Yu J. (2002) Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347, 975–982. [DOI] [PubMed] [Google Scholar]
- Hilton B. J.; Wolkowicz R. (2010) An assay to monitor HIV-1 protease activity for the identification of novel inhibitors in T-cells. PloS one 5, e10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaber R.; Majerle A.; Jerala R.; Bencina M. (2013) Noninvasive high-throughput single-cell analysis of HIV protease activity using ratiometric flow cytometry. Sensors 13, 16330–16346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella L. N.; Biswas P.; Yates M. V.; Mulchandani A.; Chen W. (2014) Quantitative assessment of in vivo HIV protease activity using genetically engineered QD-based FRET probes. Biotechnol. Bioeng. 111, 1082–1087. [DOI] [PubMed] [Google Scholar]
- Puertas M. C.; Buzon M. J.; Ballestero M.; Van Den Eede P.; Clotet B.; Prado J. G.; Martinez-Picado J. (2012) Novel two-round phenotypic assay for protease inhibitor susceptibility testing of recombinant and primary HIV-1 isolates. J. Clin. Microbiol. 50, 3909–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Cong L.; Lodato S.; Kosuri S.; Church G. M.; Arlotta P. (2011) Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 29, 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A.; Lohmueller J. J.; Silver P. A.; Armel T. Z. (2012) Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 40, 7584–7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev S. A.; Thomsen E. R.; Cieplak P.; Chudin E.; Cheltsov A. V.; Chee M. S.; Kozlov I. A.; Strongin A. Y. (2012) New details of HCV NS3/4A proteinase functionality revealed by a high-throughput cleavage assay. PloS One 7, e35759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K.; Ma L.; Han X.; Liang H.; Wei P.; Liu Y.; Lai L. (2005) The substrate specificity of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 329, 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang M. J.; Li Z.; Lim H. A.; Ng F. M.; Then S. W.; Wee J. L.; Joy J.; Hill J.; Chia C. S. (2013) A P2 and P3 substrate specificity comparison between the Murray Valley encephalitis and West Nile virus NS2B/NS3 protease using C-terminal agmatine dipeptides. Peptides 52C, 49–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.