

Abstract
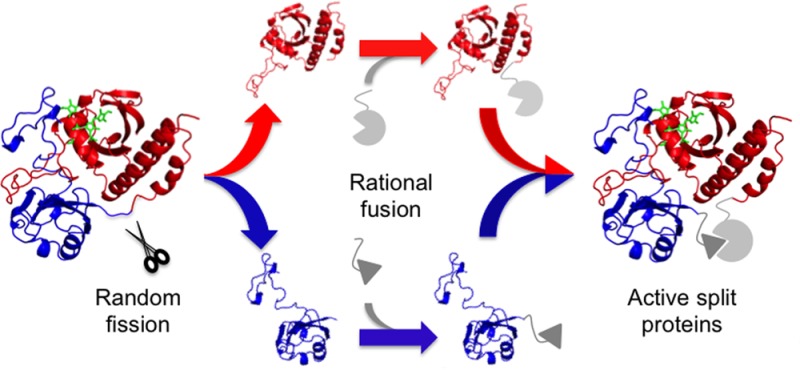
Gene fission can convert monomeric proteins into two-piece catalysts, reporters, and transcription factors for systems and synthetic biology. However, some proteins can be challenging to fragment without disrupting function, such as near-infrared fluorescent protein (IFP). We describe a directed evolution strategy that can overcome this challenge by randomly fragmenting proteins and concomitantly fusing the protein fragments to pairs of proteins or peptides that associate. We used this method to create libraries that express fragmented IFP as fusions to a pair of associating peptides (IAAL-E3 and IAAL-K3) and proteins (CheA and CheY) and screened for fragmented IFP with detectable near-infrared fluorescence. Thirteen novel fragmented IFPs were identified, all of which arose from backbone fission proximal to the interdomain linker. Either the IAAL-E3 and IAAL-K3 peptides or CheA and CheY proteins could assist with IFP fragment complementation, although the IAAL-E3 and IAAL-K3 peptides consistently yielded higher fluorescence. These results demonstrate how random gene fission can be coupled to rational gene fusion to create libraries enriched in fragmented proteins with AND gate logic that is dependent upon a protein–protein interaction, and they suggest that these near-infrared fluorescent protein fragments will be suitable as reporters for pairs of promoters and protein–protein interactions within whole animals.
Keywords: gene fission, gene fusion, mutagenesis, near-infrared fluorescent protein, protein fragment complementation, protein−protein interaction
Component limitations arise in synthetic biology because a restricted number of biological parts can be reliably used to program cellular behaviors.1,2 Gene fragmentation represents a simple strategy to overcome component limitations by converting proteins encoded by individual genes into proteins encoded by two or more gene fragments.1,2 Protein fragments that arise from fission display “AND” gate logic when the ORFs encoding the different fragments are placed under distinct transcriptional regulation so that protein fragment complementation occurs only when the promoter driving transcription of the first ORF is active AND the promoter controlling transcription of the second ORF is also active.3 Unfortunately, we often cannot predict a priori where genes (and proteins) can be fragmented into pieces to construct AND gates. In these cases, laboratory evolution can be used to identify which of the many possible fission sites within a protein are least disruptive to function.4 This combinatorial approach has the advantage that it samples all of the possible designs for constructing fragmented proteins and provides information on the backbone fission sites that yield fragments with the strongest functional complementation. In addition, comprehensive information on protein tolerance to fission can be used to guide the construction of more complex logic gates, such as three-input AND gates.5,6
Fragmented proteins often display decreased activity compared with that of their natural counterparts encoded by single polypeptides, which can limit their utility for synthetic biology applications.7 One way to create fragmented proteins with improved activity is to introduce nondisruptive backbone fission sites into homologues having enhanced stability, such as a thermophilic homologue of a fragmented mesophilic protein.8 Highly stable proteins can also be used as starting points for laboratory evolution experiments to increase the fraction of fragmented proteins that retain function within a combinatorial library,9 provided that a highly stable homologue is available as a starting point for laboratory evolution. An alternative way to enhance the cooperative function of protein fragments is to fuse protein fragments to a pair of proteins that form a stable complex and promote complementation.10 Assisted protein-fragment complementation has been used to create a variety of genetically encoded devices whose molecular outputs report on a protein–protein interaction, including devices that generate metabolic,11 visual,12−14 transcriptional,15−17 proteolytic,18,19 and antibiotic resistance20,21 outputs. The design of fragmented proteins that report on molecular interactions can be arduous,21−23 and no laboratory evolution methods have been described to simplify the discovery of protein fragments that require assistance for complementation.
We recently described a transposon mutagenesis approach for constructing libraries of vectors that express different fragmented variants of a protein.9 Libraries created using this method can be coupled to screens and selections to convert any protein into a genetically encoded AND gate, provided that one or more pairs of fragments derived from that protein cooperatively function. Because previous studies have shown that interacting proteins can assist with the complementation of protein fragments,10 we sought to explore whether this library approach could be extended so that it generates each possible two-fragment variant of a protein as fusions to a pair of proteins that associate to form a complex (Figure 1). We hypothesized that fragmented proteins mined from this type of library would require associating proteins for maximal activity. We focused our efforts on the near-infrared fluorescent protein (IFP), a mutant of the Deinococcus radiodurans bacteriophytochrome BphP that displays spectral properties that are compatible with whole body imaging in animals.24 This protein uses biliverdin (BV) as a chromophore and folds into a structure containing a knot.25,26 The extent to which IFP can be fragmented to create bimolecular fluorescence complementation assays has not been explored using laboratory evolution, although fragmented IFP have been discovered through rational design that require assistance from a protein–protein interaction for fragment complementation.27,28
Figure 1.
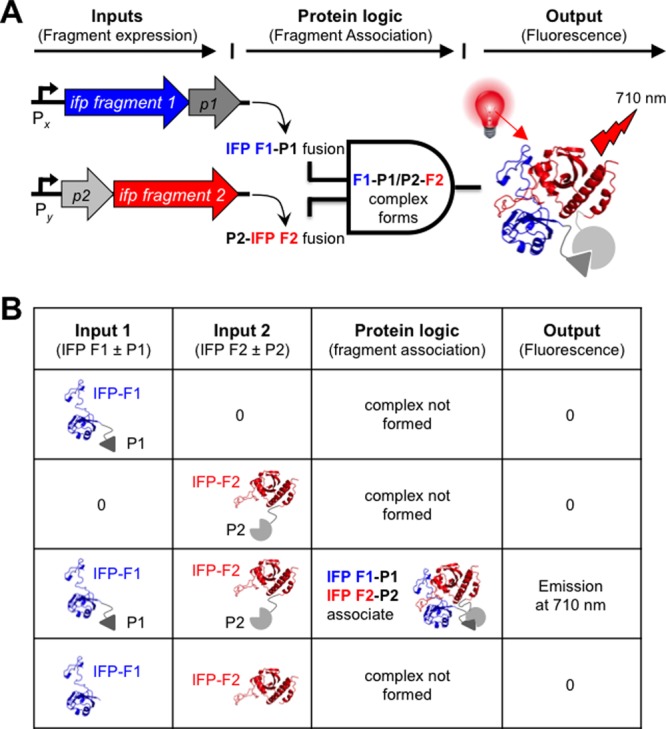
Fragmented IFP created by random fission and rational fusion. (A) Transposon mutagenesis creates clones that use two promoters (Px and Py) to control the expression of IFP fragments (F1 and F2) fused at their termini to a pair of proteins (P1 and P2) that associate. When both fusion proteins (F1–P1 and P2–F2) are expressed, they associate to form a folded two-fragment IFP that binds BV and emits in the near-infrared. (B) Truth table for a two-fragment protein AND gate that requires fusion to associating proteins for an output.
To better understand IFP tolerance to fragmentation and the best architectures for building bimolecular near-infrared fluorescence complementation devices, we developed a simple method to construct libraries encoding the different possible fragmented IFP as fusions to pairs of associating peptides (and proteins) and screened for IFP that display near-infrared fluorescence. We examined whether the fluorescent fragmented IFP required assistance from protein–protein interactions for complementation, and we investigated how the biochemical properties of fragmented IFP differed from those of full-length IFP.
Results and Discussion
Combining IFP Fission with Fusion to Peptides
To establish where IFP can be fragmented into polypeptides that cooperatively fold without assistance, we first created a library of vectors that express fragmented IFP using transposon mutagenesis9 and screened this library for variants that fluoresce like IFP in the near-infrared upon expression within Escherichia coli. Screening this library did not identify any two-fragment IFP that retained fluorescence, suggesting that the IFP structure is easily disrupted by fragmentation. Because previous studies have shown that interacting proteins can assist with the complementation of some fragmented proteins,10 we hypothesized that combining random IFP fission with fusion to associating proteins would enrich the library in variants whose fragments associate into an IFP-like structure, bind the BV chromophore, and fluoresce in the near-infrared.
To test the combined effects of random fission and rational fusion on IFP fluorescence, we constructed a library that fuses a pair of peptides (IAAL-E3 and IAAL-K3) designed to associate strongly (KD = 70 nM) to the IFP fragment termini.30 In this EK library, the IAAL-E3 peptide was added to the C-terminus of the IFP fragment that precede the backbone fission site, whereas the IAAL-K3 peptide was added to the N-terminus of the IFP fragment that follows the fission site. The IAAL-E3 and IAAL-K3 peptides were chosen for initial library construction because they are small (21 residues each), form a heterodimeric coiled-coil,30 and have been previously used to assist with fragment complementation.8 Structural studies have shown that the N- and C-termini of IAAL-E3 and IAAL-K3 that become fused to IFP fragment termini have a physical separation of ∼30 Å.31 To minimize IFP structural disruption upon fusion of fragments to termini having this separation, 12 residue glycine-rich linkers were used for library construction.
IFP Tolerates Fission When Fused to Peptides
Screening 1760 clones from the EK library identified multiple colonies with λem > 700 nm, and sequencing vectors from these colonies identified seven unique fragmented IFP having their peptide backbone cleaved distal (>25 residues) from the termini. Mapping the locations of these backbone cleavage sites onto the D. radiodurans BphP chromophore-binding domain,25 the protein that was mutated to create IFP,24 revealed that all of the fission sites are proximal to the linker that connects the PAS (Per/ARNT/Sim) and GAF (cGMP phosphodiesterase/adenyl cyclase/FhlA) domains within IFP (Figure 2A). These fission sites occur at the end of the second β-sheet within the PAS domain (117-EK and 118-EK), the end of the third β-sheet within the PAS domain (129-EK and 131-EK), and the beginning of first helix within the GAF domain (140-EK, 142-EK, and 144-EK).
Figure 2.
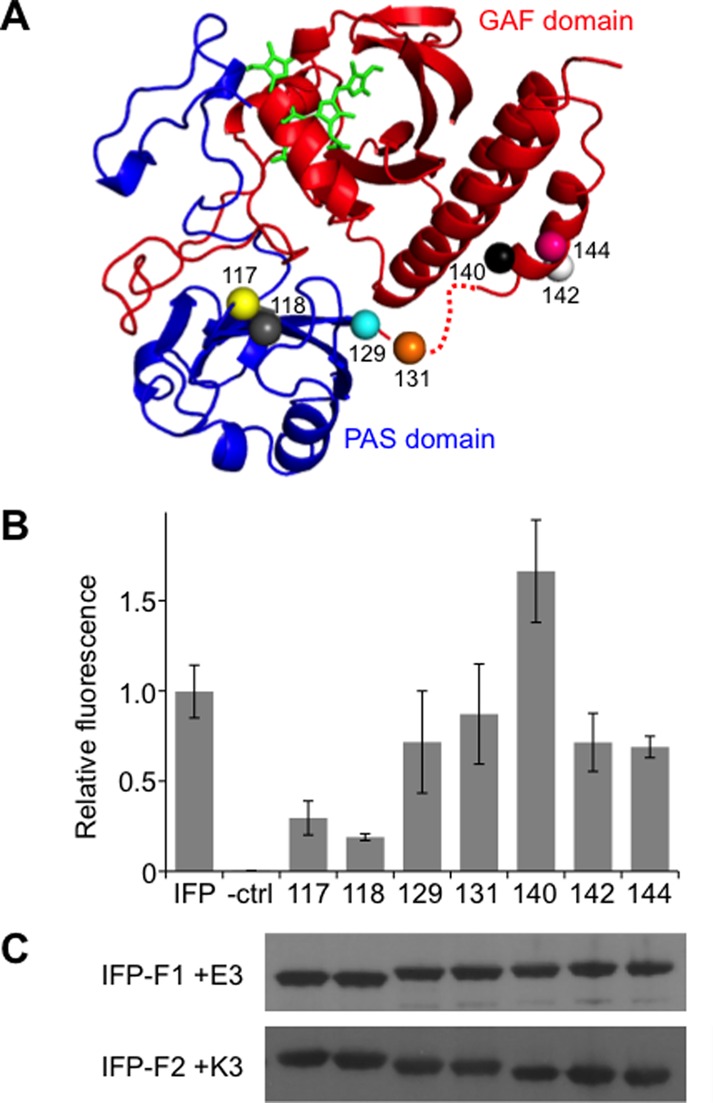
Fluorescent fragmented IFP from the EK library. (A) For those variants that displayed near-infrared fluorescence, the IFP backbone fission sites (spheres) were mapped onto the PAS (blue) and GAF (red) domains of BphP, PDB = 1ZTU,25 using PyMol. Each site represents the last IFP residue in the N-terminal fragment. The domain linker that is disordered in the BphP structure is shown as a dashed line. (B) Fluorescence of each fragmented IFP upon expression in E. coli is shown relative to that of cells expressing full-length IFP and lacking an IFP (−ctrl). Whole cell fluorescence (λex = 684 nm; λem = 710 nm) was measured at 37 °C and normalized to cell density. The fluorescence intensity obtained with cells expressing each split variant was significantly different from cells lacking an IFP (two tailed t-test; p < 0.01). (C) Western blot detection of N-terminal IFP fragments fused to IAAL-E3 (IFP-F1 + E3), which have a GST tag, and the C-terminal IFP fragments fused to IAAL-K3 (IFP-F2 + K3), which have an HA tag. Error bars represent ±1σ calculated using three or more replicates.
The finding that peptide fusion was required to discover fragmented IFP with near-infrared fluorescence suggested that these proteins may be marginally stable and display fluorescence that is sensitive to increasing temperature. To test this idea, we compared the near-infrared fluorescence of each variant with full-length IFP at 23 and 37 °C. A comparison of E. coli expressing each fragmented IFP revealed similar emission intensity as that of full-length IFP when fluorescence was analyzed at 23 °C (Figure S1). In contrast, greater variability was observed when measurements were performed at 37 °C (Figure 2B), the temperature where IFP biosensors must function to be useful within animal models. Two fragmented IFP displayed <30% of the IFP emission intensity (117-EK and 118-EK), one displayed higher fluorescence (140-EK), and all others displayed intermediate signals. To determine whether the varying fluorescence intensities of the mutants arose because they had peak emission and excitation wavelengths that differed from IFP,24 we measured the fluorescence spectrum of each variant. All of the fragmented IFP displayed maximal excitation and emission within 4 nm of intact IFP (Table S1).
To investigate whether the variability in whole cell fluorescence arose because some fragmented IFP required a different concentration of supplemental BV for maximal fluorescence, we evaluated the dependence of whole cell fluorescence on the amount of BV added to the cells expressing each fragmented IFP (Figure S2). In all cases, the concentration of BV required for half-maximal fluorescence in whole cells (2.3–15.5 μM) was lower than the concentration (80 μM) used for screening the libraries. The amounts of BV required for half-maximal fluorescence with fragmented IFP were also similar to the amount (11.2 μM) required for half-maximal fluorescence with IFP. Furthermore, the concentrations required for half maximal fluorescence did not correlate with the relative emission from each fragmented IFP. These findings suggest that the variability in emission does not arise because fragmented IFP differ in the fraction of protein that contains bound BV. To test whether the variation in emission of the IFP variants arose because the different fragment pairs accumulated to varying steady-state levels, we used western blot analysis to evaluate IFP fragment stability (Figure 2C). These experiments revealed that the different IFP fragments accumulated to similar extents when expressed from the same promoters and ribosomal binding sites (RBS) and did not reveal a correlation between near-infrared emission and protein fragment expression. Taken together, these findings suggests that the differences in fluorescence arise because of structural and/or fragment association differences among the split IFP.
The IFP BV chromophore makes a large number of noncovalent interactions with residues within the C-terminal GAF domain that reside within the C-terminal fragments. This clustering of BV-interacting residues suggested that fragmented IFP may require BV association with only one fragment to fluoresce. To test this idea, we cloned each pair of IFP gene fragments into vectors that use different inducible promoters (PT5 and PBAD) to control expression (Figure 3) and examined whether the maximum signal required expression of both the N- and C-terminal fragments. Whole cell fluorescence measurements revealed that each of the fragmented IFP displayed the highest fluorescence when E. coli harboring these vectors was grown in the presence of both arabinose and IPTG. 140-EK displayed the highest fluorescence, similar to that observed with the constructs discovered in the EK library, and similar whole cell fluorescence as cells expressing full-length IFP from either inducible promoter (data not shown). In all cases, we detected a low level of near-infrared fluorescence with cells grown only in the presence of arabinose. This trend was interpreted as arising from basal expression of the fragments under PT5 promoter regulation, which has been observed previously with the vectors used for the measurements.32 These results show that all seven of the fragmented IFP exhibit AND gate genetic logic.
Figure 3.

Two-input transcriptional regulation of fragmented IFP. The near-infrared fluorescence of E. coli (XL1-Blue) transformed with vectors that use IPTG- and arabinose-inducible promoters to express the N- and C-terminal fragments of IFP, respectively. The individual and combined effects of 5 mM arabinose (ARA) and 5 mM IPTG on whole cell fluorescence (λex = 684 nm; λem = 710 nm) was measured at 37 °C, normalized to cell density, and reported relative to the fragmented IFP with the largest signal. Error bars represent ±1σ.
Fragmented IFPs Require Assistance for Fluorescence
The paucity of fluorescent proteins discovered when IFP was subjected to fission alone suggested that the fragmented IFP mined out of the EK library might require assistance from the IAAL-E3 and IAAL-K3 coils for maximal fluorescence. This idea was tested by analyzing the fluorescence of cells expressing each fragmented IFP without fusion to these associating peptides. All of the fragmented IFP lacking peptides displayed <10% of the whole cell fluorescence observed with full-length IFP (Figure 4A) and lower fluorescence than that of identical IFP fragments fused to the IAAL-E3 and IAAL-K3 peptides. Among the different fragmented IFPs that fluoresce, peptide removal led to a loss of fluorescence that varied between 7- and 40-fold (Figure 4B).
Figure 4.
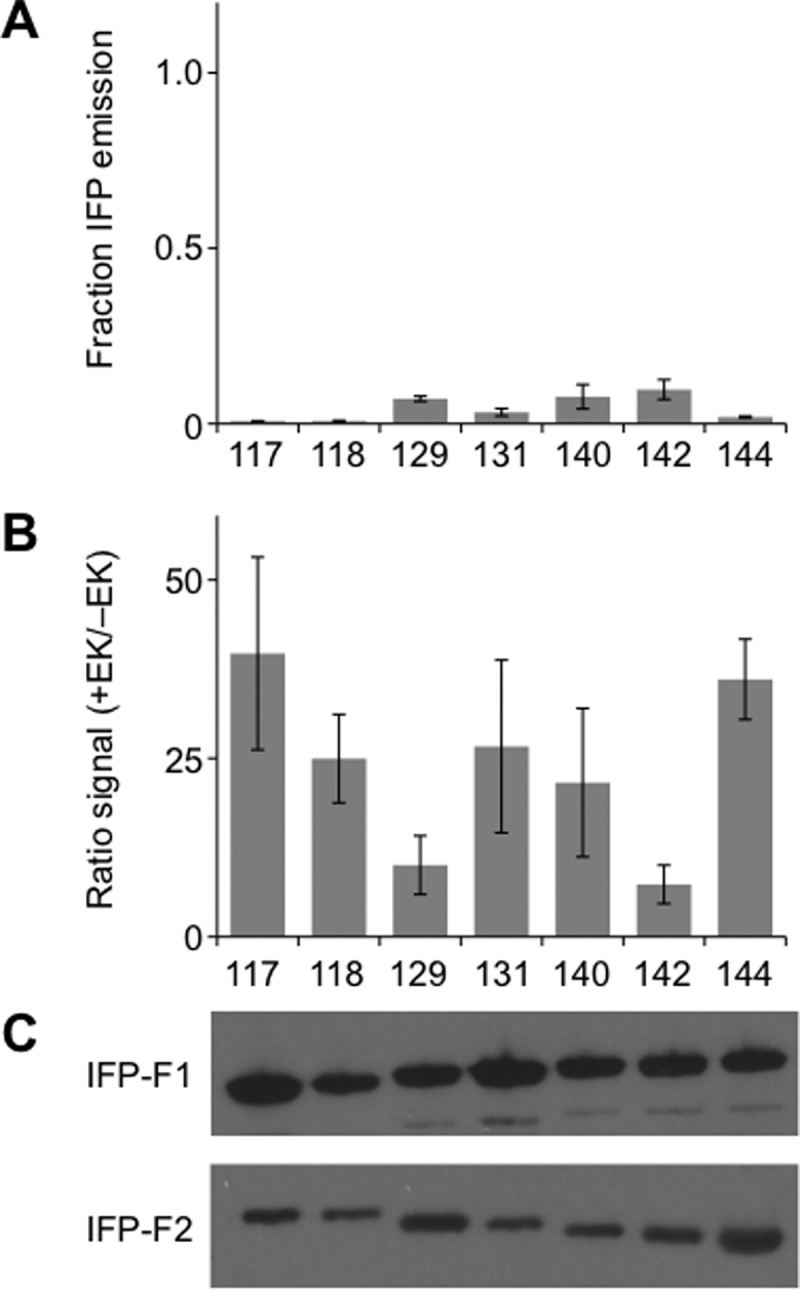
IAAL-E3 and IAAL-K3 peptide removal diminishes IFP fragment complementation. (A) Whole cell fluorescence (λex = 684 nm; λem = 710 nm) of fragmented IFP lacking peptide fusions is shown as the fraction of signal observed with cells expressing full-length IFP. Fluorescence was measured at 37 °C and normalized to cell density. (B) Ratio of fluorescence measured for two-piece IFP peptide fusions (+EK) to homologous fragmented IFP lacking peptides (−EK). Removal of the EK coils from each split IFP led to a significant change in the fluorescence intensity of each variant (two tailed t-test; p < 0.01 for all variants except 129, which had a p < 0.02). (C) Western blot detection of IFP fragments expressed in E. coli without IAAL-E3 and IAAL-K3 peptides. N-terminal IFP fragments (IFP-F1) were detected using an antibody against their GST tag, and C-terminal fragments (IFP-F2) were detected using an antibody against their HA tag. Error bars represent ±1σ.
To investigate why peptide removal decreased fluorescence of the fragmented IFP, we analyzed the fluorescence spectra, fluorescence dependence on BV, and steady-state expression of fragments lacking peptide fusions. Fragmented IFP lacking peptides displayed similar excitation and emission maxima as that of homologous variants fused to the IAAL-E3 and IAAL-K3 peptides (Table S1). In contrast, higher concentrations of BV (25.3–76.4 μM) were required for half-maximal fluorescence with IFP fragments lacking the IAAL-E3 and IAAL-K3 peptides (Figure S3). This finding indicates that these variants displayed only a fraction of their maximal whole cell fluorescence under the conditions in which library screening was performed (80 μM BV). Western blot analysis revealed that the different IFP fragments lacking peptides accumulated to similar extents (Figure 4C) and did not reveal a strong correlation between near-infrared emission and the relative expression of the different fragmented variants lacking fusion to peptides. A comparison of a subset of the fragmented IFP containing IAAL-E3 and IAAL-K3 and homologous fragmented IFP lacking these peptides (Figure S4), however, revealed that removal of the peptides from the fragmented IFP altered steady-state levels of each homologous polypeptide fragment. Removal of IAAL-E3 and IAAL-K3 increased accumulation of the N-terminal IFP fragments preceding the peptide backbone cleavage site and decreased accumulation of the C-terminal fragments following the cleavage site.
We posited that the decreased expression of C-terminal fragments upon IAAL-K3 removal arose in part because translation initiation of this fragment was decreased by changing the context of the RBS driving expression. This idea was tested by using a thermodynamic model to calculate the relative translation initiation rates from the RBS controlling IFP fragment expression.33 All seven of the C-terminal fragments had lower calculated translation initiation rates upon removal of the IAAL-K3 peptide (Figure S5). To test this prediction, we created vectors that expressed all of the IFP variants with only one fragment (C-terminal) fused to a peptide (IAAL-K3). Thermodynamic calculations predicted that all of these vectors would initiate translation of the IFP fragments to the same extent as that of fragmented IFP fused to both IAAL-E3 and IAAL-K3. We found that fragmented IFP fused to only one peptide displayed 1–7.5% of the whole cell fluorescence observed with full-length IFP (Figure 5A). This fluorescence emission was similar to or lower than that observed with homologous IFP lacking both peptides (Figure 5B). We also found that the excitation and emission spectrum of each variant was comparable to that of IFP fragments fused to IAAL-E3 and IAAL-K3 (Table S1). Western blot analysis did not reveal a strong correlation between near-infrared emission and the relative expression of the different N- and C-terminal fragments (Figure 5C). However, we found that the C-terminal fragments fused to the IAAL-K3 peptide accumulated to greater levels than the same IFP fragments lacking the peptide (Figure S4), as predicted from our RBS strength analysis.
Figure 5.

Removal of IAAL-E3 decreases IFP fragment complementation. (A) Fluorescence of E. coli (λex = 684 nm; λem = 710 nm) expressing fragmented IFP with their C-terminal fragments fused to IAAL-K3 and their N-terminal fragments lacking a peptide. Fluorescence is reported as the fraction of the signal observed with full-length IFP. (B) Ratio of fluorescence measured for fragmented IFP having IAAL-K3 fused at the terminus of one fragment (+K) to fragmented IFP homologues lacking peptide fusions (−K). The fluorescence intensity of split variants containing one K coil was not significantly different from that of variants lacking both coils, with the exception of variants 142 and 144 (two tailed t-test; p < 0.05). (C) Western blot detection of N-terminal IFP fragments (IFP-F1), which have a GST tag, and the C-terminal fragments fused to IAAL-K3 (IFP-F2 + K3), which have an HA tag. Error bars represent ±1σ.
To test whether other interacting proteins could support the complementation of IFP fragments identified in the EK library, we created vectors that expressed each of the fragmented IFP as fusions to Thermotoga maritima CheA (P2 domain) and CheY. CheA and CheY were chosen because they associate to form a heterodimer34 and have been used to assist with fragment complementation of other proteins.35 We hypothesized that CheA and CheY would be able to substitute for IAAL-E3 and IAAL-K3 without disrupting the fragmented IFP fluorescence because the distance between the termini of CheA and CheY (∼30 Å) is similar to the distance between the IAAL-E3 and IAAL-K3 termini fused to IFP fragments.31,34
We found that IFP fragments fused to CheA and CheY displayed between 15 and 71% of the whole cell fluorescence observed with full-length IFP (Figure 6A). CheA and CheY enhanced IFP fragment complementation to varying extents over IFP fragments lacking fusions to interacting proteins, ranging from 4- to 22-fold (Figure 6B). The relative effects of CheA and CheY on fragmented IFP emission were also similar to the effects of IAAL-E3 and IAAL-K3. IFP fragmented after residues 117 and 144 displayed the largest fluorescence enhancement upon fusion to the associating proteins, whereas IFP fragmented after residues 129 and 142 consistently exhibited the smallest enhancement. On average, however, fragmented IFP fused to CheA and CheY displayed lower whole cell fluorescence than that of the same IFP fragments fused to IAAL-E3 and IAAL-K3 (Figure 6C). These differences were not interpreted as arising from changes in spectral properties or fragment expression. Fragmented IFP fused to CheA and CheY displayed spectra with similar excitation and emission maxima as that of IFP (Table S1), and IFP fragments accumulated to a similar level when fused to CheA/CheY and IAAL-E3/IAAL-K3 (Figure S4). Instead, these differences are thought to arise from the varying strength of the protein–protein interactions used to assist with IFP fragment complementation. The fragment complementation correlated with the relative affinities of the CheA/CheY (KD = 200 nM) and IAAL-E3/IAAL-K3 (KD = 70 nM) complexes.30,34
Figure 6.
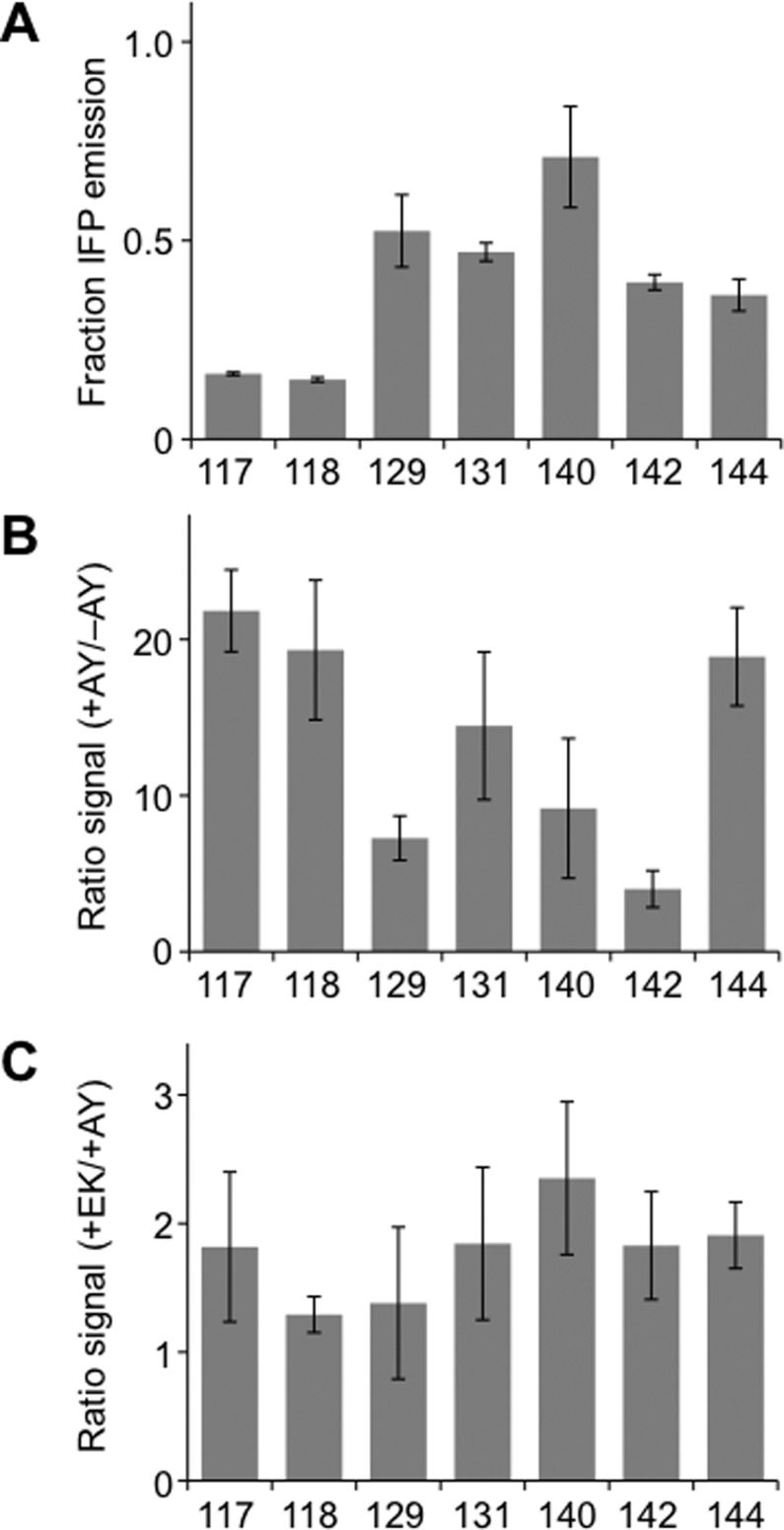
CheA and CheY rescue IFP fragment complementation. (A) Whole cell fluorescence of E. coli expressing each pair of IFP fragments as fusions to CheA and CheY. Fluorescence emission (λex = 684 nm; λem = 710 nm) measured at 37 °C was normalized to cell density and is shown relative to the signal from cells expressing full-length IFP. (B) Ratio of fluorescence measured for fragmented IFP having CheA and CheY fused at their termini (+AY) to that of homologous IFP fragments lacking CheA and CheY (−AY). The fluorescence intensity obtained with each split variant +AY was significantly different from that for homologous variants −AY (two tailed t-test; p < 0.005). (C) Ratio of fluorescence for fragmented IFP having IAAL-E3 and IAAL-K3 fused at their termini (+EK) to that for homologous IFP fragments having CheA and CheY fused at their termini (+AY). Error bars represent ±1σ. Four variants (118, 140, 142, and 144) displayed significantly higher fluorescence +EK compared with that +AY (two tailed t-test; p < 0.05).
Combining CheA/CheY Fusion with IFP Fission
We next sought to determine if combining random IFP fission with fusion to CheA and CheY would yield distinct fragmented IFP from those discovered in the EK library or fragmented IFP with a higher fluorescence. We hypothesized that screening for fragmented IFP from a library created using an alternative protein–protein interaction might identify different fragmented IFP because of differences in fragment expression, stability, and association. To test these ideas, we created a library where CheA and CheY were fused to the termini of each randomly fragmented IFP. Like the EK library, screening 1760 clones from this AY library for variants with near-infrared fluorescence yielded multiple variants with λem > 700 nm. Sequencing vectors from these cells identified nine IFP that were fragmented distal from the termini (>25 residues). Although three of these variants (118-AY, 131-AY, and 140-AY) were identified when screening the EK library, six of the variants had distinct fragmentation sites. Mapping the locations of these sites onto the BphP structure25 revealed that they cluster near the boundary of the PAS and GAF domains (Figure 7A). Two of the backbone fission sites are after the first β-strand of the PAS domain (108-AY and 111-AY), one is within the linker that connects the domains (135-AY), two are within the first helix of the GAF domain (141-AY and 143-AY), and one is after the first helix of the GAF domain (147-AY).
Figure 7.
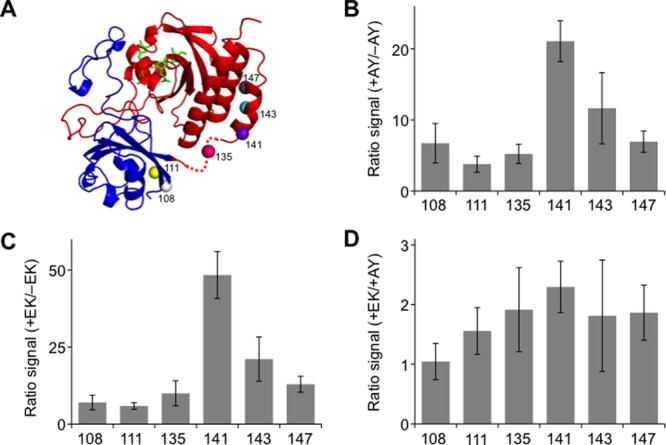
Fluorescent fragmented IFP discovered in the AK library. (A) Backbone fragmentation sites (spheres) of fragmented IFP discovered in the AY library mapped onto the PAS (blue) and GAF (red) domains of BphP25 using PyMol. (B) Ratio of fluorescence measured for fragmented IFP having CheA and CheY fused at their termini (+AY) to that for homologous variants lacking CheA and CheY (−AY). The fluorescence intensity obtained +AY was significantly different from that for homologous variants −AY (two tailed t-test; p < 0.01 for all variants excluding 143, which had a p < 0.02). (C) Ratio of fluorescence measured for fragmented IFP having IAAL-E3 and IAAL-K3 fused at their termini (+EK) to that for variants lacking peptides (−EK). The fluorescence intensity obtained +EK was significantly different from that −EK (two tailed t-test; p < 0.01). (D) Ratio of fluorescence measured for fragmented IFP having IAAL-E3 and IAAL-K3 fused at their termini (+EK) to that for variants fused to CheA and CheY (+AY). Three variants (111, 141, and 147) displayed significantly higher fluorescence +EK compared with that +AY (two tailed t-test; p < 0.05). Error bars represent ±1σ.
Cells expressing the variants mined out of the AY library displayed only a fraction (16–69%) of the fluorescence signal obtained with cells expressing full-length IFP (Figure S6). The IFP variants arising from backbone cleavage proximal to the residues linking the PAS and GAF domains showed the highest whole cell fluorescence, similar to that observed with fragmented IFP discovered in the EK library. Analysis of the emission and excitation spectrum of each variant also revealed maximal excitation and emission similar to that of IFP (Table S1). We posited that the fluorescence of fragmented IFP discovered in the AY library would depend on fusion to interacting proteins, like the variants discovered from the EK library. To test this idea, we built constructs that expressed each of these fragmented IFP without fusion to any proteins, and we measured the near-infrared fluorescence of cells transformed with these vectors. All of these cells expressing fragmented IFP displayed decreased fluorescence upon removal of CheA and CheY (Figure 7B), as observed with variants discovered in the EK library. The fluorescence of cells expressing IFP fragments fused to CheA and CheY was 4- to 21-fold higher than that for cells expressing IFP fragments lacking CheA and CheY fusions. This finding provides additional evidence that random protein fission can be combined with rational fusion to discover near-infrared fluorescence protein fragments that report on protein–protein interactions.
To establish if the variants discovered in the AY library were dependent upon CheA and CheY for maximal fluorescence, we substituted the IAAL-E3 and IAAL-K3 peptides for CheA and CheY, respectively, and analyzed IFP fragment complementation. Substitution of the peptides for CheA and CheY enhanced fragment complementation over that observed with fragmented IFP lacking fusion to any peptides or proteins (Figure 7C). As observed with fragmented IFP discovered in the EK library, fluorescence complementation was enhanced to the greatest extent when IFP fragments were fused to the IAAL-E3 and IAAL-K3 peptides (Figure 7D). These findings suggest that the affinity of the protein–protein interaction used for library construction contributes to protein-fragment complementation because the higher affinity interaction (IAAL-E3/IAAL-K3) consistently yielded stronger complementation with the fragmented IFP.
Comparison with Previous Protein Fragmentation Studies
The results described herein show how combining random gene fission with fusion to ORFs encoding associating proteins can aid the discovery of protein fragments that cooperatively function (fluorescence) and display two-input AND gate logic (two-promoter regulation). The EK and AY libraries yielded fragmented IFP that displayed near-infrared fluorescence, while fluorescent variants were not discovered in the library encoding IFP that had been subjected to random fission alone. The fragmented IFP discovered in the EK and AY libraries provide positional information on the combined effects of fission and fusion on IFPs retention of near-infrared fluorescence. All of the IFP backbone fission sites discovered using laboratory evolution were distal from the BV-binding site, suggesting that IFP fission and fusion near this chromophore binding site is disruptive to fragment complementation. The backbone fragmentation sites described here are also distinct from those evaluated in previous studies that used rational design to sample a smaller number of backbone cleavage sites within IFP28 and iRFP,27 an IFP homologue that displays higher affinity for biliverdin. These previous studies each found that backbone cleavage sites within the interdomain linker can be used to generate split proteins whose association depends upon fusion to interacting proteins. Our sites clustered proximal to the best site (132) identified in the IFP study and within the secondary structures (α-helixes, β-sheet, and loops) adjacent to this domain linker. Whether or not any of our split IFP will have any advantages or disadvantages over the rationally designed IFP/iRFP fragment complementation assays will require additional experiments that directly compare the background signal (self-association), reversibility, and fluorescence signal of each split IFP upon fusion to different interacting proteins.
The decreased near-infrared fluorescence consistently observed when the associating peptides and proteins were removed from the 13 fragmented IFP is interpreted as arising because each of these IFPs requires assistance from interacting proteins for fragment complementation. Evidence for this idea comes from the finding that IAAL-E3 removal diminished fluorescence complementation dramatically (10- to 70-fold) while only having minor effects on IFP fragment levels. These observations suggest that these fragmented IFP will be useful for reporting on protein–protein interactions within animal models, which are hard to study using fragmented GFP.36−38 The longer wavelengths of light used to excite IFP penetrates tissues to a greater extent than the wavelengths used to excite GFP family members.24 The fragmented IFP identified by laboratory evolution will also be useful for studies examining the spatial and temporal distribution of promoter activities within different cells within animals. Such measurements are currently needed to guide proteomic studies that use two-input AND gates to control metabolic labeling of newly synthesized proteins32 and to perform proteomic measurements with improved temporal and spatial resolution.39 IFP fragment complementation can be used to report on the subset of cells within an organism (bacterial or eukaryotic) where a given pair of promoters is active, information that cannot be obtained using pairs of near-infrared fluorescent protein reporters because their excitation and emission spectra overlap.40
Among the three libraries screened for protein fragments displaying near-infrared fluorescence, only the IAAL-E3/IAAL-K3 and CheA/CheY libraries yielded fragmented IFP with strong signals. The dependence on associating peptides (or proteins) for discovery of fragmented IFP can be contrasted with past fission studies. Using laboratory evolution, larger numbers of nondisruptive backbone fragmentation sites have been discovered within Thermotoga neapolitana adenylate kinase,9E. coli methionyl tRNA synthetases,32 and T7 phage RNA polymerase.5 We believe that the paucity of fragmented IFP that retained strong near-infrared fluorescence without assistance from interacting proteins arose because our IFP screen could only reliably identify fragmented proteins with near-native function, while previous studies used assays that could detect a fraction of parental protein activity. In addition, IFP fragmentation could decrease its affinity for BV, which is required for fluorescence, and the stability of IFP under the screening conditions. Support for the latter idea comes from the observation that full-length IFP displays almost 4-fold lower whole cell fluorescence when expressed at 37 °C compared with that at 23 °C (data not shown).
Future Applications of Random Fission and Rational Fusion
Our findings suggest that the best way to apply transposon mutagenesis to the discovery of other fragmented proteins whose complementation depends on a protein–protein interaction is to (i) initially screen for fragmented proteins that retain function upon fusion to a pair of interacting proteins like IAAL-E3 and IAAL-K3 and (ii) rescreen for loss-of-function upon removal of only one of the fused proteins. Testing the dependence of protein-fragment complementation by removing only one of the interacting peptides simplifies interpretation of results by avoiding changes in the translation initiation rates of the fragments. Future applications of this transposon mutagenesis approach are expected to simplify the discovery of fragmented protein devices that couple the function of other fragmented proteins to protein–protein interactions.10 In cases where fused proteins have an interaction that is conditional, e.g., dependent upon ligand11 or cofactor41 binding, fragmented proteins will display three-input AND gate logic. The first two inputs will be the promoters controlling protein fragment expression, while a third input will be the post-translational event that promotes the protein–protein interaction.
The protein fission strategy described here is expected to be useful for synthetic biology applications that involve DNA minivectors (and minicircles), small vectors that contain only the DNA sequence of interest, e.g., promoter, RBS, open reading frame, 3′ UTR, and poly A tail. These small DNAs are being developed for gene therapy because of their ability to enter difficult-to-transfect mammalian cells and survive shear forces often associated with DNA delivery.42−44 The beneficial properties of these small DNAs are inversely correlated with DNA length, and only a limited number of natural proteins can be encoded by a single minivector at the sizes (<1000 base pairs) where these small DNAs display the greatest stabilities. The benefits of minivectors could be leveraged with larger proteins if those proteins were broken into smaller fragments that fit into minivectors having sizes <1000 base pairs because pairs or trios of minivectors encoding these fragments could be delivered to difficult-to-transfect mammalian cells to reconstitute a functional protein through fragment complementation. Because our results show that IFP can be fragmented into pieces that associate through the fusion of small coils, we expect that this method will be useful for breaking natural proteins into pieces that can be encoded by minivectors.
The transposon mutagenesis approach used to randomly fragment IFP and fuse it to interacting proteins differs from a combinatorial protein engineering method previously described for this type of mutagenesis, which uses incremental truncation for random fission and blunt ligations for rational fusion.21 In contrast to transposon mutagenesis,9 incremental truncation requires the creation of two vector libraries: one that encodes the different N-terminal protein fragments and a second that encodes the C-terminal protein fragments.22 Genes encoding a pair of interacting proteins are then subcloned into each of these libraries using blunt ligation, and the two vector ensembles are cotransformed into cells and screened or selected for pairs of complementing fragments. We expect that transposon mutagenesis will be easier for many synthetic biology laboratories to implement because transposon-mediated fission does not require time-dependent sampling like incremental truncation.22 In addition, gene fragments created using transposon mutagenesis can be fused to genes encoding associating proteins by simply creating an insert encoding a pair of interacting proteins (Figure S7) and cloning that insert into the NotI restriction site created by transposon insertion (Figure S8). The sequence diversity created by transposon mutagenesis [= protein length – 1] is also more constrained than that created by incremental truncation [= (protein length – 1)×(protein length – 1)]. This occurs because transposon mutagenesis cleaves a gene into two fragments within the context of a single vector by inserting a unique NotI restriction site at different locations. In contrast, incremental truncation generates two vector libraries encoding the different types of proteins fragments and screens all possible N-terminal fragments with all possible C-terminal fragments by crossing the two libraries.21 Whether or not the more limited diversity created by transposon mutagenesis is more or less advantageous than that created by incremental truncation will require further experiments. The major advantage of creating limited diversity is that additional targeted diversity can be included in combinatorial libraries, e.g., within the linker region, and more thoroughly sampled using screens whose throughput is limited.
Materials and Methods
Library Construction
A library of randomly fragmented ifp genes was constructed as illustrated in Figure S8 using pGEX2TK-IFP1.4, a pGEX-2TK-derived vector that expresses IFP1.424 from a tac promoter with glutathione S-transferase (GST) fused to its N-terminus and a hemagglutinin (HA) tag fused to its C-terminus. A transposon containing NotI restriction sites proximal to both ends was inserted into pGEX2TK-IFP1.4 by incubating 310 ng of pGEX2TK-IFP1.4, 100 ng of M1-KanR transposon (Thermo Scienctific), and 1 U of HyperMu MuA transposase (Epicenter Biotechnologies) in a 20 μL reaction containing HyperMu buffer for 14 h at 37 °C. Reactions were terminated by adding HyperMu Stop Solution (1 μL) and incubating each reaction at 70 °C for 10 min. Total DNA was purified, electroporated into E. coli, spread onto multiple Luria Broth (LB)-agar plates containing 25 μg/mL kanamycin, and grown at 37 °C overnight. Total plasmid DNA was purified from colonies obtained on plates to obtain the Mu-insertion library. The ensemble of purified vectors was digested using restriction enzymes (BamHI and EcoRI) that cut at sites flanking the IFP gene. Then, agarose electrophoresis was used to purify the ifp-transposon hybrids (2.1 kb) away from the other DNA fragments (1 kb ifp alone, 5 kb vector backbone, and 6.1 and 7.2 kb vector backbones containing one or two 1.1 kb transposons). The ifp-transposon hybrids were cloned back into pGEX2TK to create a size-selected library (pGEX2TK-IFP-SS). DNA inserts shown in Figure S7 (f1-kanR, ek-kanR, and ay-kanR) were subcloned in place of the transposon within pGEX-IFP-SS to create three different libraries of vectors that express randomly fragmented IFP, including (i) the split IFP library that expresses fragmented IFP without fusion to other proteins, (ii) the EK library, which expresses the N- and C-terminal IFP fragments as fusions to the IAAL-E3 and IAAL-K3 peptides, and (iii) the AY library, which expresses the N- and C-terminal IFP fragments as fusions to T. maritima CheA (P2 domain; residues 175–264) and CheY. All steps involving bacterial transformations used MegaX DH10B competent cells (Life Technologies) and yielded lawns of colonies (>50 000 per transformation) whose numbers were always >10-fold larger than the number of variants in each library. Each library contained similar sequence diversity (1924 variants), which is determined by the product of the number of sites in the IFP gene (962) where NotI is inserted by the transposase MuA and the number of orientations (2) that each synthetic DNA (f1-kanR, ek-kanR, and ay-kanR) can be integrated the NotI sites introduced into the IFP gene by MuA.
Screening
E. coli Rosetta 2 DE3 (Novagen) or BL21 Star DE3 (Life Technologies) transformed with each library were spread onto LB-agar plates containing 100 μg/mL ampicillin and incubated overnight at 37 °C. Single colonies from these plates were arrayed into 96-well deep well plates containing 200 μL of LB and 100 μg/mL ampicillin and grown for 18 h at 37 °C while shaking at 250 rpm. Stationary phase cultures were diluted 4× by adding LB containing 100 μg/mL ampicillin. After growing for 1 h at 37 °C, isopropyl β-d-1-thiogalactopyranoside (IPTG) and BV (Frontier Scientific) were added to final concentrations of 0.5 mM and 80 μM, respectively. Deep well plates were incubated at 23 °C while shaking at 250 rpm for 18 h in the dark. A fraction (150 μL) of each culture was transferred to clear polystyrene 96-well flat bottom plates (Corning), cells were pelleted by centrifuging plates at 3000g for 5 min, supernatant containing excess BV was removed, and whole cell fluorescence (λex = 684 nm; λem = 695–720 nm) was measured using a Tecan M1000 plate reader. IFP variants with fluorescence (703 to 714 nm) that was >3σ higher than the signal obtained from cells lacking the IFP gene were sequenced and given names that correspond to the IFP residue at the end of the first fragment followed by letters that describe the proteins fused to the termini created by fragmentation, where EK designates IAAL-E3 and IAAL-K3 and AY designates CheA and CheY.
Fluorescence Analysis
Vectors encoding fragmented IFP were transformed into E. coli BL21 Star DE3, and individual colonies were used to inoculate LB cultures containing 100 μg/mL ampicillin. After 16 h at 37 °C and 250 rpm, cells (1 mL) were harvested by centrifugation and used to inoculate a fresh 5 mL LB culture containing 0.5 mM IPTG, 80 μM BV, and 100 μg/mL ampicillin. Cells were grown for 5 h at the indicated temperatures and 250 rpm in the dark, washed with 25% glycerol (1 mL), and resuspended in 25% glycerol (1 mL). Whole cell absorbance (600 nm) and fluorescence (λex = 684 nm, λem = 695–800 nm) were acquired from samples arrayed in flat bottom 96-well plates using a Tecan M1000 plate reader. Emission data was normalized to absorbance in each well, and data reported represent the average of three or more colonies for each sample with four replicates measured for each within 96-well plates. The vector that expresses full-length IFP (pGEX2TK-IFP1.4) and the same vector lacking the IFP gene were used as frames of reference for fragmented IFP signals. The BV dependence of whole cell fluorescence was measured using an identical protocol except that the concentration of BV was varied.
Vector Construction
To create pairs of vectors for analyzing regulated expression of each fragmented IFP, N-terminal IFP fragments were cloned into pQE80ΔcmR, a plasmid with an IPTG-inducible T5 promoter, and C-terminal IFP fragments were cloned in place of T7 RNA polymerase in the plasmid pTara,29 which has an arabinose-inducible PBAD promoter. pQE80ΔcmR was created by deleting the chloramphenicol resistance gene from pQE80 (Qiagen). All other expression vectors were generated by subcloning different NotI-flanked DNA inserts in place of the NotI-flanked inserts used to build vectors in each library.
Western Blotting
E. coli expressing the different fragmented IFP were grown as described for fluorescent analysis, harvested by centrifugation, and resuspended to identical optical densities. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out under reducing conditions using NuPAGE 12% Bis-Tris Gels (Life Technologies) and MOPS SDS running buffer and transferred to Protran nitrocellulose membrane (Whatman) using a TE 22 mini Tank Transfer Unit (GE Healthcare). After washing the nitrocellulose paper in TBST buffer (100 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween-20) for 5 min and blocking for 1 h with 10% dry milk in TBST, the membranes were incubated for 1 h with either GST rabbit (Millipore) or hemagglutinin Ab-1 (NeoMarkers) polyclonal antibodies at dilutions of 1:10000 in TBST. The nitrocellulose was then incubated for 1 h in TBST with secondary antibody, goat anti-rabbit IgG conjugated to peroxidase conjugate (Calbiochem), at a dilution of 1:10000. Signals were detected using the ECL western blotting substrate (GE Healthcare) according to the manufacturer’s protocol.
Acknowledgments
This work was supported by the National Science Foundation [1150138] (to J.J.S.), the Robert A. Welch Foundation [C-1614] (to J.J.S.), the John S. Dunn Collaborative Research Award (to A.W.M., L.Z., and J.J.S.), and the National Institutes of Health [R01 AI054830] (to L.Z.). The authors would like to thank Shirley Liu for her assistance with technical support.
Supporting Information Available
Table S1: Emission and excitation properties of fragmented IFP. Figure S1: Fluorescence of fragmented IFP at 23 °C. Figure S2: Effect of BV concentration on the fluorescence of fragmented IFP fused to IAAL-E3 and IAAL-K3 peptides. Figure S3: Effect of biliverdin concentration on the fluorescence of fragmented IFP lacking fusion to any peptides or proteins. Figure S4: Effect of changing protein fusion on the levels of IFP fragments within cells. Figure S5: Calculated translation initiation rates for C-terminal fragments containing or lacking fusion to IAAL-K3. Figure S6: Fragmented IFP discovered in the AY library. Figure S7: Synthetic DNA used for the last step of library construction. Figure S8: The method for creating libraries of fragmented proteins fused to different pairs of associating proteins. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Grünberg R.; Serrano L. (2010) Strategies for protein synthetic biology. Nucleic Acids Res. 38, 2663–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheschi R. J.; Gronenberg L. S.; Liao J. C. (2013) Protein engineering for metabolic engineering: current and next-generation tools. Biotechnol. J. 8, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shis D. L.; Bennett M. R. (2013) Library of synthetic transcriptional AND gates built with split T7 RNA polymerase mutants. Proc. Natl. Acad. Sci. U.S.A. 110, 5028–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier M.; Nixon A. E.; Shim J. H.; Benkovic S. J. (1999) Combinatorial protein engineering by incremental truncation. Proc. Natl. Acad. Sci. U.S.A. 96, 3562–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall-Shapiro T. H.; Meyer A. J.; Ellington A. D.; Sontag E. D.; Voigt C. A. (2014) A “resource allocator” for transcription based on a highly fragmented T7 RNA polymerase. Mol. Syst. Biol. 10, 742–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S.; Nguyen H. B.; Pedelacq J.-D.; Koraïchi F.; Chaudhary A.; Ganguly K.; Lockard M. A.; Favre G.; Terwilliger T. C.; Waldo G. S. (2013) A new protein–protein interaction sensor based on tripartite split-GFP association. Sci. Rep 3, 2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbaum J. J.; Schimmel P. (1991) Assembly of a class I tRNA synthetase from products of an artificially split gene. Biochemistry 30, 319–324. [DOI] [PubMed] [Google Scholar]
- Nguyen P. Q.; Liu S.; Thompson J. C.; Silberg J. J. (2008) Thermostability promotes the cooperative function of split adenylate kinases. Protein Eng., Des. Sel. 21, 303–310. [DOI] [PubMed] [Google Scholar]
- Segall-Shapiro T. H.; Nguyen P. Q.; Santos; Dos E. D.; Subedi S.; Judd J.; Suh J.; Silberg J. J. (2011) Mesophilic and hyperthermophilic adenylate kinases differ in their tolerance to random fragmentation. J. Mol. Biol. 406, 135–148. [DOI] [PubMed] [Google Scholar]
- Michnick S. W.; Ear P. H.; Manderson E. N.; Remy I.; Stefan E. (2007) Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discovery 6, 569–582. [DOI] [PubMed] [Google Scholar]
- Remy I.; Michnick S. W. (1999) Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proc. Natl. Acad. Sci. U.S.A. 96, 5394–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F.; Charlton C. A.; Blau H. M. (1997) Monitoring protein–protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc. Natl. Acad. Sci. U.S.A. 94, 8405–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker K. E.; Smith M. C. P.; Luker G. D.; Gammon S. T.; Piwnica-Worms H.; Piwnica-Worms D. (2004) Kinetics of regulated protein–protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. U.S.A. 101, 12288–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.-D.; Kerppola T. K. (2003) Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21, 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S.; Song O. (1989) A novel genetic system to detect protein–protein interactions. Nature 340, 245–246. [DOI] [PubMed] [Google Scholar]
- Russ W. P.; Engelman D. M. (1999) TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. U.S.A. 96, 863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G.; Pidoux J.; Ullmann A.; Ladant D. (1998) A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson N.; Varshavsky A. (1994) Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 91, 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. J.; Puhl H. L.; Ikeda S. R. (2009) Rapid modification of proteins using a rapamycin-inducible tobacco etch virus protease system. PLoS One 4, e7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A.; Primeau M.; Trudeau L.-E.; Michnick S. W. (2002) Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat. Biotechnol. 20, 619–622. [DOI] [PubMed] [Google Scholar]
- Paschon D. E.; Patel Z. S.; Ostermeier M. (2005) Enhanced catalytic efficiency of aminoglycoside phosphotransferase (3′)-IIa achieved through protein fragmentation and reassembly. J. Mol. Biol. 353, 26–37. [DOI] [PubMed] [Google Scholar]
- Paschon D. E.; Ostermeier M. (2004) Construction of protein fragment complementation libraries using incremental truncation. Methods Enzymol. 388, 103–116. [DOI] [PubMed] [Google Scholar]
- Choe W.; Chandrasegaran S.; Ostermeier M. (2005) Protein fragment complementation in M.HhaI DNA methyltransferase. Biochem. Biophys. Res. Commun. 334, 1233–1240. [DOI] [PubMed] [Google Scholar]
- Shu X.; Royant A.; Lin M. Z.; Aguilera T. A.; Lev-Ram V.; Steinbach P. A.; Tsien R. Y. (2009) Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science 324, 804–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. R.; Brunzelle J. S.; Forest K. T.; Vierstra R. D. (2005) A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 438, 325–331. [DOI] [PubMed] [Google Scholar]
- Wagner J. R.; Zhang J.; Brunzelle J. S.; Vierstra R. D.; Forest K. T. (2007) High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J. Biol. Chem. 282, 12298–12309. [DOI] [PubMed] [Google Scholar]
- Filonov G. S.; Verkhusha V. V. (2013) A near-infrared BiFC reporter for in vivo imaging of protein–protein interactions. Chem. Biol. 20, 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchekanda E.; Sivanesan D.; Michnick S. W. (2014) An infrared reporter to detect spatiotemporal dynamics of protein–protein interactions. Nat. Methods 11, 641–644. [DOI] [PubMed] [Google Scholar]
- Wycuff D. R.; Matthews K. S. (2000) Generation of an AraC-araBAD promoter-regulated T7 expression system. Anal. Biochem. 277, 67–73. [DOI] [PubMed] [Google Scholar]
- Litowski J. R.; Hodges R. S. (2002) Designing heterodimeric two-stranded alpha-helical coiled-coils. Effects of hydrophobicity and alpha-helical propensity on protein folding, stability, and specificity. J. Biol. Chem. 277, 37272–37279. [DOI] [PubMed] [Google Scholar]
- Lindhout D. A.; Litowski J. R.; Mercier P.; Hodges R. S.; Sykes B. D. (2004) NMR solution structure of a highly stable de novo heterodimeric coiled-coil. Biopolymers 75, 367–375. [DOI] [PubMed] [Google Scholar]
- Mahdavi A.; Segall-Shapiro T. H.; Kou S.; Jindal G. A.; Hoff K. G.; Liu S.; Chitsaz M.; Ismagilov R. F.; Silberg J. J.; Tirrell D. A. (2013) A genetically encoded AND gate for cell-targeted metabolic labeling of proteins. J. Am. Chem. Soc. 135, 2979–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H. M.; Mirsky E. A.; Voigt C. A. (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.-Y.; Beel B. D.; Simon M. I.; Bilwes A. M.; Crane B. R. (2004) In different organisms, the mode of interaction between two signaling proteins is not necessarily conserved. Proc. Natl. Acad. Sci. U.S.A. 101, 11646–11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. Q.; Silberg J. J. (2010) A selection that reports on protein–protein interactions within a thermophilic bacterium. Protein Eng., Des. Sel. 23, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.-D.; Chinenov Y.; Kerppola T. K. (2002) Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798. [DOI] [PubMed] [Google Scholar]
- Wilson C. G. M.; Magliery T. J.; Regan L. (2004) Detecting protein–protein interactions with GFP-fragment reassembly. Nat. Methods 1, 255–262. [DOI] [PubMed] [Google Scholar]
- Kerppola T. K. (2009) Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem. Soc. Rev. 38, 2876–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuet K. P.; Tirrell D. A. (2013) Chemical tools for temporally and spatially resolved mass spectrometry-based proteomics. Ann. Biomed. Eng. 42, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova D. M.; Verkhusha V. V. (2013) Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 10, 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff K. G.; aCuller S. J.; Nguyen P. Q.; McGuire R. M.; Silberg J. J.; Smolke C. D. (2009) In vivo fluorescent detection of Fe–S clusters coordinated by human GRX2. Chem. Biol. 16, 1299–1308. [DOI] [PubMed] [Google Scholar]
- Zhao N.; Fogg J. M.; Zechiedrich L.; Zu Y. (2011) Transfection of shRNA-encoding minivector DNA of a few hundred base pairs to regulate gene expression in lymphoma cells. Gene Ther. 18, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanese D. J.; Fogg J. M.; Schrock D. E.; Gilbert B. E.; Zechiedrich L. (2012) Supercoiled minivector DNA resists shear forces associated with gene therapy delivery. Gene Ther. 19, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenler S.; Wiklander O. P.; Badal-Tejedor M.; Turunen J.; Nordin J. Z.; Hallengärd D.; Wahren B.; Andaloussi S. E.; Rutland M. W.; Smith C. E.; Lundin K. E.; Blomberg P. (2014) Micro-minicircle gene therapy: implications of size on fermentation, complexation, shearing resistance, and expression. Mol. Ther.–Nucleic Acids 2, e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.