

Abstract
Bionanocapsules (BNCs) are hollow nanoparticles consisting of hepatitis B virus (HBV) envelope L proteins and have been shown to deliver drugs and genes specifically to human hepatic tissues by utilizing HBV-derived infection machinery. The complex of BNCs with liposomes (LPs), the BNC–LP complexes (a LP surrounded by BNCs in a rugged spherical form), could also become active targeting nanocarriers by the BNC function. In this study, under acidic conditions and high temperature, BNCs were found to fully fuse with LPs (smooth-surfaced spherical form), deploying L proteins with a membrane topology similar to that of BNCs (ie, virosomes displaying L proteins). Doxorubicin (DOX) was efficiently encapsulated via the remote loading method at 14.2%±1.0% of total lipid weight (mean ± SD, n=3), with a capsule size of 118.2±4.7 nm and a ζ-potential of −51.1±1.0 mV (mean ± SD, n=5). When mammalian cells were exposed to the virosomes, the virosomes showed strong cytotoxicity in human hepatic cells (target cells of BNCs), but not in human colon cancer cells (nontarget cells of BNCs), whereas LPs containing DOX and DOXOVES (structurally stabilized PEGylated LPs containing DOX) did not show strong cytotoxicity in either cell type. Furthermore, the virosomes preferentially delivered DOX to the nuclei of human hepatic cells. Xenograft mice harboring either target or nontarget cell-derived tumors were injected twice intravenously with the virosomes containing DOX at a low dose (2.3 mg/kg as DOX, 5 days interval). The growth of target cell-derived tumors was retarded effectively and specifically. Next, the combination of high dose (10.0 mg/kg as DOX, once) with tumor-specific radiotherapy (3 Gy, once after 2 hours) exhibited the most effective antitumor growth activity in mice harboring target cell-derived tumors. These results demonstrated that the HBV-based virosomes containing DOX could be an effective antitumor nanomedicine specific to human hepatic tissues, especially in combination with radiotherapy.
Keywords: drug delivery system, liposomes, bionanocapsule, doxorubicin, targeting, chemoradiotherapy
Introduction
Nanoparticles (NPs) including liposomes (LPs), polymers, and micelles have been expected as promising drug delivery system (DDS) nanocarriers for cancer chemotherapy.1 Based on the mechanism of NPs for recognizing target cells, tissues, and organs, they are classified into the following two groups. “Passive targeting-based NPs” mainly utilize the enhanced permeability and retention (EPR) effect, which is observed in tumors showing active angiogenesis or inflamed sites. Because of the relatively leaky vasculature and poor lymphatic drainage of macromolecules, NPs with a size of ~100 nm could be spontaneously extravasated from vessels and accumulate in tumors or inflamed areas.2 “Active targeting-based NPs” display targeting molecules on their surface and thereby deliver therapeutic agents specifically to the pathological areas. For example, NPs modified with folic acid or transferrin were shown to accumulate efficiently in tumors expressing receptors for each ligand.3,4 However, in vivo, active targeting of cancer cells did not always show higher therapeutic efficacy than in passive targeting.5 This is because the endothelial lining is not very leaky, and there are a large number of pericytes or smooth muscle cells covering the tumor blood vessels. In addition, the high tumor cell density, dense matrix hindering penetration, and high interstitial fluid pressure contribute to decreased efficiency. Especially, in case of ~10 nm-sized NPs, while active targeting could contribute to efficient early binding to tumor blood vessels (within 4 hours), passive targeting showed more efficient retention within the tumors after 24 hours.6 On the other hand, it has been recently revealed that EPR effect is hardly observed in the human body.5,7 Thus, the indications of therapeutics using passive targeting-based NPs are limited. These situations have led us to combine chemotherapy using active targeting-based NPs with other types of therapies (eg, sonotherapy, radiotherapy, phototherapy, thermotherapy, and magnetic therapy).
The hepatitis B virus (HBV) is a small DNA virus displaying three types of envelope proteins embedded in lipid bilayers.8 The largest envelope L protein was found to be indispensable for the maturation of the virus9 and for the interaction with an unidentified HBV receptor, which is necessary for the initial step of human hepatic cell-specific infection.10,11 The in vivo behavior of HBV has led us to assume that HBV is a naturally occurring human liver-specific nanocarrier. When the L protein gene was expressed in Saccharomyces cerevisiae, NPs with a size of ~50 nm were overexpressed in the cells12 and could be promptly purified by heat treatment and column chromatography.13,14 The NP consists of about 110 molecules of L protein embedded in a unilamellar LP, displaying human liver-specific receptor outwardly.14 When the various mammalian cells were exposed to NPs, they were able to attach onto the cell surface in a human hepatic cell-specific manner and then enter the cells at the same rate as HBV.15 Following the encapsulation of chemicals, nucleic acids, or proteins by electroporation, NPs could deliver payloads specifically to human hepatic cells not only in vitro but also in vivo.16 Thus, it was demonstrated that NPs (hereafter designated as bionanocapsules [BNCs]) are novel active targeting nanocarriers harboring HBV-derived infection machinery. Next, we formulated BNC–LP complexes by liposomal fusion (ie, LP surrounded by BNCs),17 incorporated doxorubicin (anticancer regent; DOX), and intravenously injected 6.0 mg/kg of DOX into xenograft mice harboring human hepatocellular carcinoma-derived tumors every 4 days.18 Owing to active targeting, the intratumoral concentration of DOX was higher than that of Doxil (structurally stabilized PEGylated LPs containing DOX). Interestingly, the BNC–LP–DOX complexes showed relatively higher stability in blood than LP–DOX complexes, suggesting that the surface modification of LPs with BNCs evaded the recognition by opsonins. However, mice treated with BNC–LP–DOX complexes unexpectedly lost weight, which was presumably due to the cytotoxicity of the drug. It is therefore a prerequisite to improve the active targeting ability of BNC–LP complexes, which could be achieved by reducing the free BNCs from the complexes.
In this study, we found that BNCs could fully fuse with LPs under acidic conditions and high temperature, deploying L proteins with a membrane topology similar to that of BNCs (ie, virosomes19 displaying L proteins). After formulating the virosomes containing DOX, we observed good antitumor activity in a mouse xenograft model at a low dose (2.3 mg/kg as DOX, two times) without the loss of body weight. Furthermore, to maximize the chemotherapeutic efficacy, we evaluated the treatment in combination with radiotherapy.
Materials and methods
Bionanocapsules
BNCs were prepared from S. cerevisiae AH22R− cells carrying the BNC expression plasmid, pGLDLIIP39-RcT,12 according to the purification protocol described previously.13 Protein concentrations were determined with a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific Inc., Rockford, IL, USA) using bovine serum albumin as a control protein. To remove contaminated materials, all samples were precipitated with ice-cold acetone containing 10% (w/v) trichloroacetic acid and 0.07% (v/v) 2-mercaptoethanol at −20°C for 1 hour. The precipitated samples were washed with ice-cold acetone and then subjected to BCA protein assay kit.
Liposomes
Dipalmitoylphosphatidylcholine (NOF Corporation, Tokyo, Japan), dipalmitoyl phosphatidylethanolamine (NOF Corporation), dipalmitoyl phosphatidylglycerol sodium salt (NOF Corporation), and cholesterol (Nacalai Tesque, Inc., Kyoto, Japan) were mixed in a round bottom flask at a molar ratio of 15:15:30:40, dissolved in a chloroform/methanol (2:1, v/v) mixture, and then evaporated at 37°C using a rotary evaporator to produce a thin hemispherical lipid film. To produce anionic LPs with a size of ~100 nm, the film was hydrated with buffer containing 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 4.0) and 120 mM (NH4)2SO4 at 60°C. The freeze–thaw cycle was repeated five times using a liquid nitrogen bath. The crude solution of LPs was subjected to a Lipex extruder (Northern Lipids, Vancouver, BC, Canada) equipped with a polycarbonate filter (pore size, 200 nm) four times at 60°C, and twice with a filter (pore size, 50 nm). The solution was further sonicated for 10 minutes on ice using an Astrason ultrasonic disruptor (Misonix, Farmingdale, NY, USA).
Virosomes containing DOX
LPs were incubated in Britton–Robinson buffer (0.1 M H3BO3, 0.1 M CH3COOH, 0.1 M H3PO4, 0.5 M NaOH, pH 3.0) at 70°C for 5 minutes, mixed with BNCs, and then kept at 70°C for 55 minutes. The solutions were subjected to a Sephadex G-25 (GE Healthcare, Buckinghamshire, UK) gel-filtration column equilibrated with 10 mM HEPES buffer (pH 7.4) containing 100 mM NaCl and 3.4% (w/v) sucrose. Total lipid concentration was estimated from the cholesterol concentration, which was determined by a Cholesterol E-Test Wako kit (Wako, Osaka, Japan) utilizing cholesterol oxidase, peroxidase, 4-aminoantipyrine, and N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3,5-dimethoxyaniline.20 To introduce DOX (Sigma-Aldrich, St Louis, MO, USA) into the virosomes by a remote loading method,21,22 360 μL of 5 mg/mL DOX-HCl solution was added to 1.4 mL of virosome solution (15 mg as total lipids, prewarmed at 60°C), and then incubated at 60°C for 20 minutes with gentle stirring. After the removal of free DOX by a Sephadex G-25 gel-filtration column equilibrated with 10 mM HEPES buffer (pH 7.4) containing 100 mM NaCl and 3.4% (w/v) sucrose, virosomes containing DOX were obtained. Following disruption with 0.1% (w/v) sodium dodecyl sulfate (SDS) and 0.1 N HCl, the total DOX concentration was calculated from the fluorescence intensity derived from DOX (excitation at 488 nm; emission at 515 nm), which was determined by a Varioskan fluorescence microplate reader (Thermo Electron, Vantaa, Finland).
Physicochemical analyses
The formation of virosomes was analyzed by CsCl isopycnic ultracentrifugation (5%–40% [w/v] CsCl, 10 mM HEPES, 120 mM (NH4)2SO4, pH 4.0) using a P40ST swing rotor (Hitachi, Tokyo, Japan) at 24,000 rpm at 25°C for 16 hours. The fractions (500 μL each) were collected from the top, of which the protein and lipid concentrations were determined by the Micro BCA protein assay kit and the Cholesterol E-Test Wako kit, respectively. The sizes and ζ-potentials of BNCs, virosomes, and virosomes containing DOX were measured in PBS (phosphate-buffered saline; 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) at 25°C by a dynamic light scattering, model Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). For observation with transmission electron microscopy (TEM), the virosomes and LPs were adsorbed onto a carbon-coated copper grid (JEOL, Tokyo, Japan), negatively stained using 2% (w/v) uranyl acetate, and subjected to TEM analysis using a TEM model JEM1011 (JEOL).
Proteinase protection assay
The virosomes (5 μg of L protein) were incubated in 50 μL of PBS containing 50 ng of tosyl phenylalanyl chloromethyl ketone-treated trypsin (Sigma-Aldrich) at 37°C for 0.5 hour, filtered with an Amicon Ultra-0.5 mL centrifugal filter (cutoff 100 kDa; Millipore Corporation, Billerica, MA, USA), subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and then stained with silver (EzStain Silver; ATTO Corp., Tokyo, Japan). Concurrently, the gel was blotted onto a nylon membrane (Immobilon-P; Millipore Corporation) and subjected to Western blotting using a mouse monoclonal anti-S antibody (IgM, clone 5124A; Institute of Immunology, Tokyo, Japan) or a mouse monoclonal anti-preS1 antibody (IgG, clone T0606; Institute of Immunology). Immunoreactive bands were visualized with an Amersham ECL prime Western blotting detection reagent (GE Healthcare).
In vitro cytotoxicity assay
Human hepatocellular carcinoma cell lines, Huh7 (RCB1366, RIKEN, Wako) and NuE (a gift from Professor Takushi Tadakuma, National Defense Medical College, Tokorozawa, Japan), and human colon carcinoma cell line WiDr (JCRB0224; JCRB Cell Bank, Osaka, Japan) were cultured in Dulbecco’s Modified Eagle’s Medium (Nacalai Tesque, Inc.) supplemented with 10% (v/v) fetal bovine serum (FBS; PAA Laboratories GmbH, Linz, Austria) at 37°C in a humidified atmosphere containing 5% (v/v) CO2. Huh7 (target of BNC) and WiDr (nontarget of BNC) cells were seeded in 96-well cell culture plates (Nalge Nunc, Naperville, IL, USA) at a density of ~5×103 cells/well, and then cultured for 24 hours. These cells were treated with DOX, DOXOVES (structurally stabilized PEGylated LPs containing DOX; Formu Max Scientific, Palo Alto, CA, USA), LP–DOX complexes, and virosomes containing DOX at appropriate concentrations (0.195–50 μg/mL as DOX) at 37°C for 6 hours. After incubation at 37°C for 24 hours in fresh medium, cell viability was measured using a commercial 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) assay reagent (Nacalai Tesque, Inc.) according to the manufacturer’s protocol. The formation of tetrazolium from WST-8 was monitored on a Varioskan fluorescence microplate reader at 450 nm.
Subcellular localization analyses
Approximately 5×103 Huh7 or WiDr cells were cultured in an eight-well glass-bottomed chamber (Nalge Nunc) at 37°C for 24 hours, incubated with 50 μg/mL (as DOX) of DOX, LP–DOX complexes, and virosomes containing DOX at 37°C for 90 minutes. After washing the cells with PBS three times, these cells were fixed with 4% (w/v) paraformaldehyde in PBS at 37°C for 20 minutes, washed three times with PBS, and then stained with Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) in PBS at 37°C for 20 minutes. The fluorescence derived from DOX (excitation at 488 nm; emission at 515 nm) and Hoechst 33342 (excitation at 350 nm; emission at 461 nm) was observed under a confocal laser scanning microscope model FV-1000D (Olympus, Tokyo, Japan). The average fluorescence intensity of each region of interest was measured using ImageJ 1.47v software (National Institute of Health, Bethesda, MD, USA).
Evaluation of chemotherapeutic efficacies
Six-week-old male BALB/c nude (nu/nu) mice (Japan SLC, Hamamatsu, Japan) were subcutaneously injected on their backs with ~106 cells (NuE or WiDr cells) in 50 μL PBS mixed with 50 μL Matrigel (BD Biosciences, Bedford, MA, USA). When the solid-tumor size reached 100 mm3 (9–10 days after injection), the mice received intravenous drug injections at 2.3 mg (as DOX)/kg (body weight) on days 0 and 5. The drugs used were DOX, DOXOVES, LP–DOX complexes, and virosomes containing DOX, each suspended in 150 μL of HEPES buffer (pH 7.4), 100 mM NaCl, and 3.4% (w/v) sucrose. Tumor volume and body weight were measured for 15 days. Based on the largest (a, mm) and smallest (b, mm) superficial diameters of each tumor, the tumor volume (V, mm3) was calculated using the following equation V = ab2/2. All experiments were performed following the protocol approved by the Institutional Animal Care and Use Committee of Nagoya University.
In vivo cytotoxicity assay
Mice were intravenously injected with drugs at 5 mg (as DOX)/kg (body weight) on days 0, 3, 7, 10, 14, 17, and 21. After blood samples and whole hearts were isolated 72 hours after the last injection, serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured by the core laboratory at the ASAN medical center to monitor liver function, and 10–20 pieces of tissue samples (1×2×2 mm) were taken from the myocardium of the left ventricle. Tissue samples were immediately fixed in 3% (v/v) glutaraldehyde in PBS. Before sectioning, tissue samples were washed with PBS, postfixed in 1% (w/v) osmium tetroxide for 1 hour, and dehydrated by passing through a series of graded ethanol. Then, tissue samples were infiltrated with acetone and poly/bed 812 plastic resin (Ted Pella, Inc., Redding, CA, USA), and embedded in plastic block molds with 100% poly/bed 812. Ultra thin sections (80 nm) were cut from sample block, and mounted on 100 mesh copper grids. Grids were stained with 2% (w/v) uranyl acetate and Reynold’s lead citrate stain. Grids were examined with a TEM model JEM1011 (JEOL).
Evaluation of chemoradiotherapeutic efficacies
Six-week-old male BALB/c nude mice were subcutaneously injected at their right hind legs with ~3×106 cells (Hep3B cells) in 50 μL PBS. When the size of the solid tumor reached from 70 to 120 mm3, the mice received intravenous drug injections at 10 mg (as DOX)/kg (body weight) on day 0. The drugs used were DOX, LP–DOX complexes, virosomes containing DOX, and virosomes alone, each suspended in 150 μL of HEPES buffer (pH 7.4), 100 mM NaCl, and 3.4% sucrose. After 2 hours, the tumors were irradiated at 3 Gy using a 6 MV photon beam linear accelerator (CL/1800; Varian Medical System, Palo Alto, CA, USA). Body weights and tumor volumes were monitored for 26 days. All experiments were performed following the protocol approved by the Institutional Animal Care and Use Committee of the ASAN Institute for Life Sciences, Seoul, Republic of Korea.
Results and discussion
Optimization of BNC–LP complex preparation
Our previous studies17,18 demonstrated that the mixtures of BNCs and LPs at a weight ratio of 1:2 or 1:20 (at a molar ratio, BNC [as particle]:LP [as lipids] =1:1.6×104 or 1:1.6×105, respectively) formed BNC–LP complexes spontaneously at room temperature under neutral conditions. However, following CsCl isopycnic ultracentrifugation analyses, the free form of BNCs or LPs was always found in the complex preparations. Since these free BNCs were considered to suppress the human hepatic cell-specific delivery by BNC–LP complexes in a competitive inhibition manner, the weight ratio of BNC:LP was investigated from 1:10 (1:8.0×104 at molar ratio) to 1:30 (1:2.4×105 at molar ratio) to reduce the unexpected generation of free BNCs. Furthermore, since BNCs are stable up to 70°C,14 the phase transition temperature of LPs is estimated to be above 70°C, and BNC contains fusogenic activity in the translocation motifs of preS2 and/or S region.23,24 We then examined whether the BNC–LP complex formation was enhanced with incubation at 70°C under acidic conditions (pH 3.0). As shown in Figure 1, narrower peaks of BNC–LP complexes were observed at 70°C, suggesting that BNC–LP complexes became homogeneous. Among the BNC–LP complexes prepared at 70°C, the highest yield of BNCs was obtained at BNC:LP =1:20 (Figure 1D). Consequently, the BNC–LP complexes prepared at BNC:LP =1:20 (1:1.6×105 at molar ratio), 70°C, and pH 3.0 were promptly used for further experiments without any purification.
Figure 1.

CsCl isopycnic ultracentrifugation analyses of BNC–LP complexes.
Notes: The mixtures of BNCs (as particle) and LPs (as lipids) at weight ratios of 1:10 (at a molar ratio of 1:8.0×104; A, B), 1:20 (at a molar ratio of 1:1.6×105; C, D), and 1:30 (at a molar ratio of 1:2.4×105; E, F) were incubated at 37°C and pH 3.0 (A, C, E) or 70°C and pH 3.0 (B, D, F) for 1 hour, and then analyzed using CsCl isopycnic ultracentrifugation (24,000 rpm, 25°C, 16 hours). Protein concentrations (red) were determined with a BCA protein assay kit. Lipid concentrations (blue) were estimated from cholesterol concentration, which was determined using a Cholesterol E-Test Wako kit. Density (green) was measured using an electronic balance. The yield (%) of BNCs in each BNC–LP complex preparation is indicated in the graphs.
Abbreviations: BNC, bionanocapsule; LP, liposome; abs, absorbance; BCA, bicinchoninic acid.
Formation of virosomes of HBV envelope L proteins
On TEM observation, the BNC–LP complexes prepared at room temperature under neutral conditions (pH 7.0) (Figure 2A, panel a) exhibited a rugged spherical structure of 112.5±19.7 nm (mean ± SD, n=10) in diameter, which agreed well with a previous report.17 The interface between BNCs and LP became obscure (Figure 2A, panel b), suggesting the occurrence of membrane fusion. When prepared at 70°C and pH 3.0, BNC–LP complexes exhibited a smooth-surfaced spherical structure of 111.4±19.6 nm (mean ± SD, n=10) in diameter (Figure 2A, panel c), which is similar to that of LPs (Figure 2A, panel d). When BNCs and BNC–LP complexes were digested with trypsin, anti-preS1 immunoreactive ~48 and ~43 kDa peptides (full-length N,O-glycosylated and N-glycosylated L protein, respectively) disappeared (Figure 2B, upper panel). Following trypsinization of BNCs, the anti-S immunoreactive peptides with molecular weights of ~16, ~19, ~26, and ~29 kDa were protected (Figure 2B, lower panel), these peptides corresponded to nonglycosylated and O-glycosylated forms of the ~16 kDa peptide (from Gly-19 in the preS2 region to Arg-129 in the S region) and ~26 kDa peptide (from Gly-19 in the preS2 region to the C-terminal in the S region), respectively.12 Similar peptides were found in the trypsinized BNC–LP complexes that strongly suggests that the membrane topology of Hepatitis B virus surface antigen (HBsAg) L protein was well conserved and the preS region was presented outside of the BNC–LP complexes. Based on the elimination of rugged structure in the BNC–LP complexes by incubation at 70°C in acidic conditions, it was postulated that HBsAg L proteins in BNCs were transferred to LPs, presumably by membrane fusion. Compared with the TEM images of previous BNC–LP complexes (Figure 2A, panel a),17 the shape was dramatically changed from a rugged spherical structure to a smooth spherical structure (Figure 2A, panel c). Since BNC contains fusogenic activity in the translocation motifs of the preS2 and/or S region,23,24 both high temperature and acidic conditions might enhance the membrane fusion between BNCs and LPs. In addition, the proteinase protection assay showed that the membrane topology of HBsAg L protein was well conserved after the fusion between BNCs and LPs. It was strongly suggested that HBsAg L proteins in BNCs were disassembled into micelles and then spontaneously integrated into LPs (Figure 3), indicating the formation of virosomes.19 While the composition of BNCs is ~10% (w/w) lipids and ~90% L proteins,25 that of virosomes is estimated to be ~96% lipids and ~4% L proteins. It was expected that virosomes can incorporate DOX by remote loading at comparable levels of LPs alone. Collectively, virosomes could be regarded as LPs transplanted with the targeting ability and fusogenic activity of HBsAg L protein, namely LPs armed with HBV-derived infection machineries.
Figure 2.

Characterization of BNC–LP complexes.
Notes: (A) Transmission electron microscopy (TEM) photographs of (a) the BNC–LP complexes prepared at room temperature under neutral conditions, (b) the interface between BNCs and LP in the boxed area of (a), White arrowheads indicate the interface between BNCs and LP. (c) The BNC–LP complexes prepared at 70°C under acid conditions (namely virosomes), and (d) LP. (B) The virosomes treated with trypsin were separated by SDS-PAGE, and then immunoblotted with anti-preS1 antibody (upper panel) and anti-S antibody (lower panel). Asterisks indicate the bands of anti-preS1 immunoreactive proteins (~48 and ~43 kDa) and anti-S immunoreactive proteins (~16, ~19, ~26, and ~29 kDa). The sizes (C) and ζ-potentials (D) of virosomes (green), virosomes containing DOX (red), and LPs (blue) were measured by DLS. Scale bars represent 100 nm.
Abbreviations: BNC, bionanocapsule; LP, liposome; Try, trypsin; Ab, antibody; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; DOX, doxorubicin; DLS, dynamic light scattering.
Figure 3.

Two forms of BNC–LP complexes.
Notes: TEM photographs of BNC–LP complexes and virosomes. Scale bars represent 100 nm.
Abbreviations: BNC, bionanocapsule; LP, liposome; RT, room temperature; TEM, transmission electron microscopy.
Remote loading of DOX into BNC–LP complexes
In previous studies, the BNC–LP–DOX complexes had been prepared by mixing LP–DOX complexes with BNCs at room temperature under neutral conditions.18 When the mixing was performed at 70°C under acidic conditions, the DOX in LPs leaked out unexpectedly. Therefore, the remote loading of DOX21,22 was applied to preformed virosomes. Based on the change in fluorescent intensity by SDS-HCl treatment, the efficiency of DOX incorporation was estimated to be 95.7%±0.7% (mean ± SD, n=6), which was comparable to that of Doxil (a structurally stabilized LPs containing DOX).26 The weight ratio of DOX in the virosomes was calculated to be 14.2%±1.0% of total lipids (mean ± SD, n=3), which was higher than those of Doxil (12.5%)26 and previous version of BNC–LP–DOX complexes (13.3%).18 The average composition, average size, and average ζ-potential of virosomes containing DOX were estimated at BNC:LP:DOX =1:20:2.8 (w/w/w) (1:1.6×105:2.2×104 at molar ratio), 118.2±4.7 nm (mean ± SD, n=5) (Figure 2C), and −51.1±1.0 mV (mean ± SD, n=5) (Figure 2D), respectively. It was considered that both the size and ζ-potential of BNC–LP–DOX complexes are ideal for systemic administration.27,28
Stability of virosomes containing DOX
The stability of virosomes containing DOX was evaluated in 10 mM HEPES buffer (pH 7.4) containing 100 mM NaCl and 3.4% sucrose (outer aqueous phase buffer) at 4°C or 37°C. Based on the relative amount of DOX remaining, the virosomes were found to retain DOX long-term (8 weeks) at 4°C rather than at 37°C (Figure 4A), while the diameter of the virosomes increased slightly (Figure 4B). Thus, the structure of virosomes containing DOX was well maintained in the outer aqueous phase buffer at 4°C for at least 8 weeks. Furthermore, the virosomes containing DOX were stored in 50% FBS (containing active complements) at 37°C for 8 days. More than 70% of the incorporated DOX was retained in the virosomes during the first 2 days, whereas about 80% of the incorporated DOX was gradually released from the virosomes during another 4 days (Figure 4C). The diameter of the virosomes containing DOX was significantly increased within 6 days (Figure 4D). When using the previous version of BNC–LP–DOX complexes, the conjugation of BNCs could enhance the retention time of DOX in the bloodstream more efficiently.18 Thus, the virosomes containing DOX would harbor comparable level of stability for use in vivo.
Figure 4.

Stability of virosomes containing DOX.
Notes: The virosomes containing DOX were kept in outer aqueous phase buffer at 4°C (red) and 37°C (blue) for 8 weeks. The relative amount of DOX (A) and particle size (B) were measured at 0, 1, 2, 3, 4, and 8 weeks. The virosomes containing DOX were kept in outer aqueous phase buffer at 4°C and 37°C for 8 weeks. The relative amount of DOX (C) and particle size (D) were measured at 0, 1, 2, 4, 5, 6, and 8 days. The virosomes containing DOX were kept in 50% FBS at 37°C for 8 days. Error bars represent standard deviation (n=3).
Abbreviations: DOX, doxorubicin; FBS, fetal bovine serum.
In vitro cytotoxicity of virosomes containing DOX
Huh7 cells (target cells) and WiDr cells (nontarget cells) were incubated with DOX, LP–DOX complexes, DOXOVES (commercial DOX-containing PEGylated LPs), and virosomes containing DOX at DOX concentrations of 0.195–50 μg/mL for 6 hours, and then subjected to WST-8 assay. In Huh7 cells, the virosomes containing DOX showed higher cytotoxicity than the LP–DOX complexes and DOXOVES (Figure 5A). The IC50 value (50% inhibitory concentration for cell growth) of each drug was ~4.0 μg/mL (DOX), ~12.5 μg/mL (virosomes containing DOX), ~25.0 μg/mL (LP-DOX), and ~46.0 μg/mL (DOXOVES). In WiDr cells (Figure 5B), while DOX showed strong cytotoxicity (~2.5 μg/mL), the other drugs showed low cytotoxicity (>100 μg/mL). These results demonstrated that the full fusion with BNCs conferred LP–DOX complexes with the targeting ability that is specific to human hepatic cells.
Figure 5.
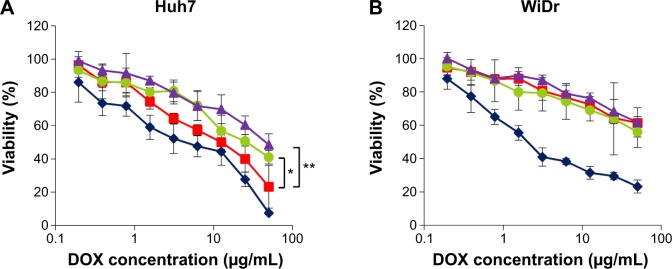
In vitro cytotoxicity assay.
Notes: Huh7 (A) or WiDr (B) cells were treated with DOX (blue diamonds), DOXOVES (purple triangles), LP–DOX complexes (green circles), and virosomes containing DOX (red squares) at 37°C for 6 hours. Cell viability was measured using a commercial WST-8 assay reagent. Error bars represent standard deviation. t-test, **P<0.01, *P<0.05.
Abbreviations: DOX, doxorubicin; LP, liposome; DOXOVES, structurally stabilized PEGylated LPs containing DOX; WST-8, 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt.
Subcellular localization of DOX
When Huh7 and WiDr cells were treated with DOX, virosomes containing DOX, and LP–DOX complexes at a DOX concentration of 50 μg/mL for 90 minutes, the subcellular localization of DOX in each cell was analyzed under a confocal laser scanning microscope (Figure 6A). Based on the DOX-derived fluorescence, free DOX itself was spontaneously and efficiently accumulated at the nucleus in both cell types, the virosomes containing DOX exhibited higher fluorescence intensity in the nuclei of Huh7 cells (~59% of free DOX) than WiDr cells (~24% of free DOX), and LP–DOX complexes showed similar fluorescence intensity in the nuclei of both cell types (~32% and ~39%, respectively) (Figure 6B). Since both human hepatic cell-specific targeting domains11 and fusogenic domains23,24 were identified in HBV envelope L protein, the virosomes could attach onto the target cells, subsequently enter the cells by either endocytosis or membrane fusion in an HBV receptor-dependent manner,29 and then release DOX into the cytoplasm. On the other hand, in the nontarget cells, the virosomes containing DOX could enter the cells less efficiently than LP–DOX complexes. Full fusion of BNCs with the LP–DOX complexes facilitated either the active incorporation of DOX in target cells or the repulsion of DOX from nontarget cells.
Figure 6.

Subcellular localization of DOX in Huh7 and WiDr cells.
Notes: (A) Huh7 or WiDr cells treated with drugs (DOX, virosomes containing DOX, LP–DOX) for 90 minutes were observed under a confocal LSM. Scale bars represent 20 μm. (B) The relative fluorescence intensity of nuclear DOX in Huh7 and WiDr cells was determined using the ImageJ program. Error bars represent standard deviation (n=7). t-test, **P<0.01.
Abbreviations: DOX, doxorubicin; LP, liposome; LSM, laser scanning microscope.
Chemotherapeutic efficacy of virosomes containing DOX
The antitumor effect of virosomes containing DOX has been evaluated by in vivo tumor growth assays using a mouse xenograft model. Mice were intravenously injected with a low dose of DOX (2.3 mg/kg) twice (day 0 and day 5). Since virosomes alone showed no significant effect on tumor growth in comparison with control, we examined the chemotherapeutic efficacies of DOX, LP–DOX complexes, virosomes containing DOX, and DOXOVES. On day 15, in mice harboring NuE (target cell)-derived tumors, the average rates of tumor growth inhibition (T/C, treated versus control; %) of DOX, LP–DOX complexes, virosomes containing DOX, and DOXOVES were 90.9%, 81.6%, 54.1%, and 38.6%, respectively (Figure 7A). Meanwhile, in mice harboring WiDr (nontarget cell)-derived tumors on day 15, the T/C values (%) of DOX, LP–DOX complexes, virosomes containing DOX, and DOXOVES were 90.6%, 82.8%, 81.7%, and 74.9%, respectively (Figure 7B). None of the mice exhibited significant weight loss during the observations (Figure 7C and D). Compared with the LP–DOX complexes, the virosomes containing DOX, even at lower dose than the previous version (2.3 mg/kg as DOX, twice versus 6.0 mg/kg as DOX, every 4 days18), showed higher antitumor activity against NuE-derived tumors, indicating that the full fusion with BNCs successfully endowed LP–DOX complexes with the human hepatic cell-specific active targeting ability. However, their antitumor activities were lower than that of DOXOVES (driven by passive targeting machinery). If the virosomes containing DOX are more stabilized in the bloodstream, the complexes could exhibit higher antitumor activity than DOXOVES by taking advantage of the passive and active targeting abilities simultaneously. Generally, severe weight loss occurred because of high concentration of free DOX in serum.26 In the previous study,18 since free BNCs might attenuate the targeting ability of BNC–LP–DOX complexes in a competitive inhibition manner, we optimized the BNC–LP ratio for reducing the amount of free BNC as much as possible. Thus, we could lower free DOX concentration in serum and thereby repress the unexpected weight loss of mice. Other possible reasons for the repression of weight loss of mice are as follows: 1) the serum half-life of DDS nanocarrier may be extended by the shape change of virosomes (from rugged to smooth structure); 2) the smooth structure of virosomes may be more suitable for the in vivo targeting and/or the cellular uptake than the rugged structure.
Figure 7.

Evaluation of chemotherapeutic efficacies.
Notes: Nude mice harboring NuE (A, C) or WiDr (B, D) xenograft tumors were intravenously injected twice (0 and 5 days) with virosomes containing DOX (red squares), LP–DOX complexes (green circles), DOX (blue diamonds), DOXOVES (purple triangles), and control (black crosses) at a low DOX dose (2.3 mg/kg). Tumor volume (A, B) and body weight (C, D) of the mice were measured for 15 days. Error bars represent standard deviation (n=5). Black arrows indicate intravenous injections.
Abbreviations: DOX, doxorubicin; LP, liposome.
Cytotoxicity of virosomes containing DOX
As for the in vivo cytotoxicity of virosomes containing DOX, since BNC possesses a HBV-derived infection system, there have been concerns that the virosomes containing DOX (the complex of BNCs and LP–DOX) could induce the immune system into attacking healthy liver cells. However, BNC (targeting molecule of virosomes) has already been shown as a safe biomaterial in humans.30,31 And, the LP–DOX complexes (ie, Doxil, DOXOVES) have also been confirmed safe in human.26 It was therefore considered that the virosomes containing DOX are not cytotoxic in vivo. Furthermore, we examined the in vivo cytotoxicity of the drug in mice. Because the virosomes can target human liver only (not mouse liver) and DOX shows strong cytotoxicity in heart,26 we examined the cardiotoxicity by observation under TEM. As shown in Figure 8, the DOX-treated mice displayed more extensive mitochondrial degeneration in association with marked swelling, cristae disorganization, and myofibril degeneration compared with nontreated mice. Mice treated with LP–DOX complexes and those with virosomes containing DOX exhibited no significant change from nontreated mice, indicating that both drugs have no cardiotoxicity. As for liver injury, the virosomes containing DOX did not increase AST and ALT levels in mouse serum compared with the levels in control mice (40±3 U/L and 22±2 U/L, respectively; n=5). Based upon the safety data in human, the cardiotoxicity, and the serum data, it was strongly suggested that the virosomes containing DOX are not cytotoxic in vivo.
Figure 8.

Evaluation of cardiotoxicity.
Notes: Mice were intravenously injected with DOX, LP–DOX complexes, and virosomes containing DOX (as DOX 5 mg/kg) two times per week for 3 weeks. Myocardial samples were isolated from the left ventricle. Mitochondria (M) and myofibril (My) structures were shown in myocardium electron micrographs. Scale bars represent 1 μm.
Abbreviations: DOX, doxorubicin; LP, liposome.
Chemoradiotherapeutic efficacy of virosomes containing DOX
Radiotherapy is one of the critical options for the treatment of various cancers, but several cancers including hepatocellular carcinoma are poorly responsive or even nonresponsive to radiotherapy. Recently, it has been proposed that the combination of chemotherapy using tumor-targeting NPs with radiotherapy could improve the therapeutic efficacy synergistically.32,33 Anticancer drugs have also acted as radiosensitizers, improving the radiotherapeutic efficacy. To examine whether exposure to ionizing radiation (IR) enhances the in vivo chemotherapeutic efficacy of virosomes containing DOX, xenograft mice bearing Hep3B (target cell)-derived tumors were treated with virosomes only, DOX, LP–DOX complexes, and virosomes containing DOX (10.0 mg/kg as DOX, once), followed by treatment with IR (3 Gy, once after 2 hours). As shown in Figure 9A, the T/C values (%) of virosomes only, DOX, LP–DOX complexes, and virosomes containing DOX on day 26 were 80.1%, 49.9%, 54.4%, and 34.3%, respectively. In combination with IR, the T/C values (%) of IR alone and that of virosomes only, DOX, LP–DOX complexes, and virosomes containing DOX on day 26 were 68.1%, 69.2%, 40.8%, 31.5%, and 23.4%, respectively (Figure 9B). The amount of time it took for the tumors to reach equal volumes in the mice treated with virosomes containing DOX plus IR (day 26) was measured. Nontreated mice (control), mice treated with IR alone, DOX plus IR, and LP–DOX complexes plus IR required 7, 15, 19, and 22 days, respectively. Notable changes in body weight were not observed in any group, indicating that severe toxicity did not occur in those mice (Figure 9C). These results demonstrated that the combination of virosomes containing DOX with IR exerted the strongest radiosensitizing effect when compared with the combination of others (DOX, LP–DOX complexes) with IR.
Figure 9.

Evaluation of chemoradiotherapeutic efficacies.
Notes: Nude mice harboring Hep3B xenograft tumors were intravenously injected once (0 day) with virosomes containing DOX (red squares), LP–DOX complexes (green circles), DOX (blue diamonds), virosomes (yellow circles), group of IR treatment alone (purple triangles), and control (black crosses) at a high DOX dose (10 mg/kg). The mice were treated without (A) or with (B) IR 2 hours later. Tumor volume (A, B) and body weight (C) of the mice were measured for 26 days. Error bars represent standard deviation (n=7). Black arrows indicate intravenous injections. Red arrows indicate IR treatment.
Abbreviations: DOX, doxorubicin; LP, liposome; IR, ionizing radiation.
Conclusion
We have established the preparation method for HBV envelope L protein-derived virosomes based on the full fusion of BNCs with LPs under acidic conditions and high temperature. The virosomes incorporated DOX efficiently via the remote loading method, resulting in physiochemical properties that were suitable for systemic administration. When human hepatic cells were exposed to the virosomes, the virosomes containing DOX entered the cells using HBV-derived infection machinery and readily released DOX into the cytoplasm. In a mouse xenograft model harboring human hepatic cell-derived tumors, the virosomes containing DOX exhibited high antitumor effects even at low dose of DOX without significant loss of body weight, which were comparable to those of commercial DOX-containing PEGylated LPs (Doxil). These effects were synergistically enhanced by radiotherapy.
Acknowledgments
We thank Mr Takuo Nakamoto and Mr Chao Cai for their technical assistance. This work was supported in part by the KAKENHI (Grant-in-Aid for Scientific Research [A] [21240052, 25242043 to SK], Grant-in-Aid for Young Scientists [B] [25870310 to MI]), the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (BRAIN) (to SK), the Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries and Food Industry (to SK), the Health Labor Sciences Research Grant from the Ministry of Health Labor and Welfare (to SK), the Korean Health Technology R&D Project, Ministry for Health and Welfare, Republic of Korea (HI06C0868 and HI10C2014, to EKC), and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (NRF-2012R1A2A2A01014671, to EKC).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):274–281. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 3.Goren D, Horowitz AT, Tzemach D, et al. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin Cancer Res. 2000;6(5):1949–1957. [PubMed] [Google Scholar]
- 4.Iinuma H, Maruyama K, Okinaga K, et al. Intracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancer. Int J Cancer. 2002;99(1):130–137. doi: 10.1002/ijc.10242. [DOI] [PubMed] [Google Scholar]
- 5.Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161(2):175–187. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 6.Kunjachan S, Pola R, Gremse F, et al. Passive versus active tumor targeting using RGD- and NGR-modified polymeric nanomedicines. Nano Lett. 2014;14(2):972–981. doi: 10.1021/nl404391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010;197:3–53. doi: 10.1007/978-3-642-00477-3_1. [DOI] [PubMed] [Google Scholar]
- 8.Heerman KH, Goldman U, Schwartz W, et al. Large surface proteins of hepatitis B virus containing the pre-s sequence. J Virol. 1984;52(2):396–402. doi: 10.1128/jvi.52.2.396-402.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruss V, Ganem D. The role of envelope proteins in hepatitis B virus assembly. Proc Natl Acad Sci U S A. 1991;88(3):1059–1063. doi: 10.1073/pnas.88.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Seyec J, Chouteau P, Cannie I, Guguen-Guillouzo C, Gripon P. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J Virol. 1999;73(3):2052–2057. doi: 10.1128/jvi.73.3.2052-2057.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neurath AR, Kent SB, Strick N, Parker K. Identification and chemical synthesis of a host cell receptor binding site on hepatitis B virus. Cell. 1986;46(3):429–436. doi: 10.1016/0092-8674(86)90663-x. [DOI] [PubMed] [Google Scholar]
- 12.Kuroda S, Otaka S, Miyazaki T, Nakao M, Fujisawa Y. Hepatitis B virus envelope L protein particles: synthesis and assembly in Saccharomyces cerevisiae, purification, and characterization. J Biol Chem. 1992;267(3):1953–1961. [PubMed] [Google Scholar]
- 13.Jung J, Iijima M, Yoshimoto N, et al. Efficient and rapid purification of drug- and gene-carrying bio-nanocapsules, hepatitis B virus surface antigen L particles, from Saccharomyces cerevisiae. Protein Expr Purif. 2011;78(2):149–155. doi: 10.1016/j.pep.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Yamada T, Iwabuki H, Kanno T, et al. Physicochemical and immunological characterization of hepatitis B virus envelope particles exclusively consisting of the entire L (pre-S1 + pre-S2 + S) protein. Vaccine. 2001;19(23–24):3154–3163. doi: 10.1016/s0264-410x(01)00017-2. [DOI] [PubMed] [Google Scholar]
- 15.Yamada M, Oeda A, Jung J, et al. Hepatitis B virus envelope L protein-derived bio-nanocapsules: mechanisms of cellular attachment and entry into human hepatic cells. J Control Release. 2012;160(2):322–329. doi: 10.1016/j.jconrel.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Yamada T, Iwasaki Y, Tada H, et al. Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol. 2003;21(8):885–890. doi: 10.1038/nbt843. [DOI] [PubMed] [Google Scholar]
- 17.Jung J, Matsuzaki T, Tatematsu K, et al. Bio-nanocapsule conjugated with liposomes for in vivo pinpoint delivery of various materials. J Control Release. 2008;126(3):255–264. doi: 10.1016/j.jconrel.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Kasuya T, Jung J, Kinoshita R, et al. Bio-nanocapsule-liposome conjugates for in vivo pinpoint drug and gene delivery. Methods Enzymol. 2009;464:147–166. doi: 10.1016/S0076-6879(09)64008-8. [DOI] [PubMed] [Google Scholar]
- 19.Kaneda Y. Virosomes: evolution of the liposome as a targeted drug delivery system. Adv Drug Deliv Rev. 2000;43(2–3):197–205. doi: 10.1016/s0169-409x(00)00069-7. [DOI] [PubMed] [Google Scholar]
- 20.Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem. 1974;20(4):470–475. [PubMed] [Google Scholar]
- 21.Lasic DD, Čeh B, Stuart MC, et al. Transmembrane gradient driven phase transitions within vesicles: lessons for drug delivery. Biochim Biophys Acta. 1995;1239(2):145–156. doi: 10.1016/0005-2736(95)00159-z. [DOI] [PubMed] [Google Scholar]
- 22.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 23.Oess S, Hildt E. Novel cell permeable motif derived from the PreS2-domain of hepatitis-B virus surface antigens. Gene Ther. 2000;7(9):750–758. doi: 10.1038/sj.gt.3301154. [DOI] [PubMed] [Google Scholar]
- 24.Rodríguez-Crespo I, Núñez E, Yélamos B, et al. Fusogenic activity of hepadnavirus peptides corresponding to sequences downstream of the putative cleavage site. Virology. 1999;261(1):133–142. doi: 10.1006/viro.1999.9823. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi M, Asano T, Utsunomiya M, et al. Recombinant hepatitis B virus surface antigen carrying the pre-S2 region derived from yeast: purification and characterization. J Biotechnol. 1988;8(1):1–21. [Google Scholar]
- 26.Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Byrne JD, Napier ME, DeSimone JM. More effective nano-medicines through particle design. Small. 2011;7(14):1919–1931. doi: 10.1002/smll.201100442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao K, Li Y, Luo J, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–3446. doi: 10.1016/j.biomaterials.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watashi K, Urban S, Li W, Wakita T. NTCP and beyond: opening the door to unveil hepatitis B virus entry. Int J Mol Sci. 2014;15(2):2892–2905. doi: 10.3390/ijms15022892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raz R, Koren R, Bass D. Safety and immunogenicity of a new mammalian cell-derived recombinant hepatitis B vaccine containing pre-S1 and pre-S2 antigens in adults. Isr Med Assoc J. 2001;3:328–332. [PubMed] [Google Scholar]
- 31.Shouval D, Roggendorf H, Roggendorf M. Enhanced immune response to hepatitis B vaccination through immunization with a pre-S1/pre-S2/S vaccine. Med Microbiol Immunol. 2015;204:57–68. doi: 10.1007/s00430-014-0374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lammers T, Hennink WE, Storm G. Tumor-targeted nanomedicines: principles and practice. Br J Cancer. 2008;99(3):392–397. doi: 10.1038/sj.bjc.6604483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lammers T, Subr V, Peschke P, et al. Image-guided and passively tumour-targeted polymeric nanomedicines for radiochemotherapy. Br J Cancer. 2008;99(6):900–910. doi: 10.1038/sj.bjc.6604561. [DOI] [PMC free article] [PubMed] [Google Scholar]