

Abstract
Photoderivatized polymer-coated gold surfaces have been developed following a perfluorophenylazide-based double ligation strategy. Gold-plated quartz crystal microbalance (QCM) crystals were initially covalently functionalized with a monolayer of poly(ethylene glycol) (PEG), using photo- or thermolytic nitrene formation and insertion. The polymer surfaces were subsequently used as substrates for photoinsertion of carbohydrate-derivatized photoprobes, yielding different recognition motifs for selective protein binding. The resulting robust and biocompatible sensor surfaces were applied to a flow-through QCM instrument for monitoring lectin–carbohydrate interactions in real time. The results clearly show the predicted lectin selectivity, demonstrating the applicability of the approach.
Selective surface generation and functionalization form the basis for intense research in a number of diverse areas such as chemical sensors, interface engineering, and nanotechnology. A versatile method to covalent derivatization is based on the specific chemistry of arylazides. Upon thermal activation or light irradiation, the azide functionality in these structures becomes converted to a highly reactive nitrene species that readily inserts into CH and NH bonds.1 Especially perfluorophenylazides (PFPAs),2 which produce markedly enhanced insertion yields, have thus been used to thermo- and photochemically introduce functional groups in a range of entities, including proteins, nanostructures, and synthetic polymers.3–9 The versatility of the PFPA chemistry makes it an attractive choice for surface modification. Surfaces can either be globally modified by the technique, or discrete areas can be addressed by arraying techniques such as photomasking and conventional printing devices.
The surface presentation of recognition motifs based on bioactive compounds is central for many applications in bioanalytical and biomaterials science, and carbohydrates and glycomimetic structures are in this context attracting increasing interest.10–14 Interactions between cellular glycans and proteins have been found to be of special importance in many biological processes, being involved in cell–cell interactions, cell communication, cell proliferation, and cell death. This emerging significance prompts for new means to analytically study these interactions. To date, various methods have been developed to analyze interactions of carbohydrates and proteins, including biosensors, enzyme-linked lectin assays (ELLAs) and cell assays, nuclear magnetic resonance, calorimetric techniques, and, more recently, microarray technologies.15–23 Among these methods, the quartz crystal microbalance (QCM) biosensor format is being increasingly adopted due to its convenient means of operation coupled with high performance.24–33 Measurements can also be performed in real time using native analytes without the need of any labeling procedures.
Efficient carbohydrate immobilization techniques to the sensor surface are a prerequisite for the preparation of biosensor systems. In principle, three general strategies can be used to enable carbohydrate presentation at sensor surfaces: direct covalent conjugation of carbohydrate structures to the gold electrode, noncovalent adsorption of carbohydrates on a modified surface, and covalent conjugation of carbohydrate-derivatives to a modified surface. Because of the flow-through QCM system used in the present study, the covalent methods are advantageous, leading to enhanced stability and durability of the surfaces. Most methods based on direct coupling to the gold surface make use of thiol- or disulfide-derivatized carbohydrate structures. These methods have proven their value, especially when combined with alkyl- and/or oligoethylene linker regions, however, to some extent leading to release of the compound from the surface due to the rather weak thiol–gold bond.34 For this reason, new and robust methods of attachment need to be developed, generating high-density, biocompatible surfaces.
This issue has been addressed in the present study. A range of carbohydrate structures was covalently immobilized to polymer-coated gold surfaces, based on the versatility of arylazide chemistry. A double surface ligation methodology was developed, and the resulting carbohydrate-presenting matrices were subjected to binding and competition assays between the carbohydrates and a panel of plant lectins.
EXPERIMENTAL SECTION
General
Commercially available starting materials for synthesis were purchased from Aldrich, Fluka, Lancaster, and Senn Chemicals. Carbohydrate-derivatized photoprobes 4–6 (Figure 1) were synthesized as previously reported.15 Concanavalin A (Con A) from Canavalia ensiformis, Ulex europaeus agglutinin-I (UEA), and Pisum sativum agglutinin (PSA), were purchased from Sigma. Viscum album agglutinin (VAA) was purified and checked for activity as described.35 Reactions were carried out with anhydrous solvents under a nitrogen atmosphere where appropriate. Chemical reactions were monitored with thin-layer chromatography using precoated silica gel 60 (0.25 mm thickness) plates (Macherey-Nagel). Flash column chromatography was performed on silica gel 60 (SDS 0.040–0.063 mm). 1H and 13C spectra were recorded with a DMX 500 instrument (Bruker) at 298 K in CDCl3 using the residual signal from CHCl3 (1H NMR, δ = 7.25 ppm; 13C NMR, δ = 77.0 ppm) as an internal standard. Quartz crystal microbalance experiments were performed using an Attana 100 instrument (Attana).
Figure 1.

Double surface ligation of carbohydrate structures to gold-plated QCM crystals. The gold electrode is initially derivatized with disulfide photoprobe 3, yielding a perfluorophenylazide-presenting surface. A poly(ethylene glycol) (PEG) monolayer is subsequently covalently attached to the surface by azide thermo- or photolysis. Finally, carbohydrate-derivatized photoprobes are coupled to the PEG-layer by the nitrene photoinsertion reaction.
2,2′-Dithioethyl Bis(4-azido-2,3,5,6-tetrafluorobenzoate) (3)
Synthesis of PFPA-disulfide 3 was performed according to Scheme 1. 4-Azido-2,3,5,6-tetrafluorobenzoic acid4 (1) (1.00 g, 4.3 mmol) was dissolved in CH2Cl2 (20 mL) at 0 °C. 2,2′-Dithiodiethanol (2) (0.33 g, 2.1 mmol), N,N′-dimethylaminopyridine (DMAP, 0.05 g, 0.43 mmol), and 1,3-dicyclohexylcarbodiimide (DCC, 0.95 g, 4.6 mmol) were added to the solution, and the mixture was allowed to warm to room temperature and stirred for 12 h. After removal of the precipitated dicyclohexylurea by filtration, the reaction mixture was recovered by extraction with CH2Cl2. The organic layer was washed with water and brine, dried over Na2SO4, and concentrated. Purification of the residue by flash column chromatography (hexane/ethyl acetate 6:1) afforded compound 3 in 40% yield (0.49 g). 1H NMR (500 MHz, CDCl3) δ 3.03 (4H, t, J = 6.5 Hz, CH2–S), 4.62 (4H, t, J = 6.5 Hz, CH2–OCO); 13C NMR (125 MHz, CDCl3) δ 37.2, 64.4, 107.7, 124.0, 139.6, 142.1, 144.6, 147.7, 159.6.
Scheme 1a.
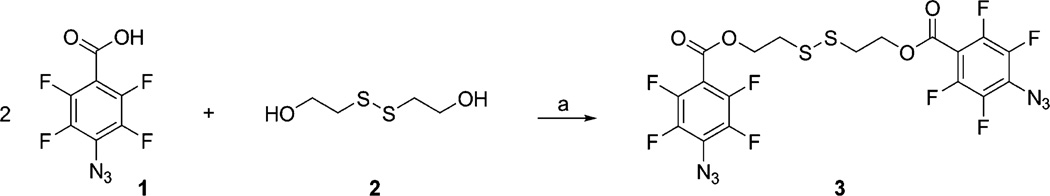
aReagents and conditions: (a) DMAP, DCC, CH2Cl2, 0 °C–rt, dark, 12 h, 40%.
Preparation of Carbohydrate-Derivatized Surfaces
Gold-plated 10 MHz quartz crystals (Attana) were initially immersed into a mixture of H2O2 (33%), NH3 (33%) and distilled water (1:1:3 v/v/v) at 80 °C for 5 min to clean the surfaces. The resulting crystals were repeatedly rinsed with distilled water and dried under a stream of nitrogen. The cleaned crystals were subsequently immersed into a solution of PFPA–disulfide 3 (14 mM in CH2Cl2) (Figure 1) at room temperature in the dark overnight, rinsed with CH2Cl2 to remove excess compound 3, and dried under a stream of nitrogen. The PFPA-activated crystals were immersed into a melt of poly(ethylene glycol) (Mr 20 000) at 70 °C, after which the temperature was increased to 140 °C for 20 min. The resulting PEG-coated crystals were withdrawn from the melt, sonicated in distilled water to remove unbound PEG, and dried under a stream of nitrogen. The dried PEG-coated crystals were subsequently immersed in solutions of PFPA-derivatized carbohydrates (10 mM in ethanol) for 5 min. After drying under nitrogen, the crystals were irradiated with a medium-pressure mercury UV-source (Hanovia) for 5 min. After UV irradiation, the crystals were finally rinsed with ethanol and then dried under nitrogen.
QCM Conditions
Protein binding to the surfaces was monitored by frequency logging with Attester 1.1 (Attana) and recorded as the resulting frequency shifts (Δf). A negative response shift is indicative of increased surface mass resulting from protein binding. The fabricated crystals were mounted in the flow-through QCM system and equilibrated with buffer solution (10 mM PBS, pH 7.4) prior to manipulations/measurements. Blocking of the surfaces was performed by injection of bovine serum albumin (BSA) (10 mg/mL in PBS) until a stable frequency baseline was obtained. A continuous flow of running buffer (PBS 10 mM, pH 7.4, 50 µL/min) was used throughout, and all test samples were prepared in the running buffer (PBS 10 mM, pH 7.4; injection volume: 50 µL). A maximum protein concentration of 10 µM was used for Con A, PSA, and UEA, whereas 5 µM was used for VAA due to its comparatively low solubility. Bound lectins were released from the surfaces between measurements by two successive injections of low pH buffer (PBS 10 mM, pH 1.5).
Estimation of KD
The apparent KD for the interaction between the α-d-mannopyranoside-functionalized surface and Con A was estimated from binding experiments up to saturation. Increasing concentrations (ranging from 5 nM to 10 µM) of Con A were injected over the QCM surface, and the frequency decrease was recorded. The software package GraphPad Prism (GraphPad) was adopted for nonlinear regression analysis determining the apparent KD using eq 1, where Δf is the frequency difference at a certain concentration, Δfmax the maximum frequency difference (maximum binding), and [L] the lectin concentration.
| (1) |
RESULTS AND DISCUSSION
In order to acquire a stable and biocompatible sensor, suitable for the QCM flow-through system, a polymer-based surface was addressed. A polymer coating of the crystal gold electrodes would produce an acceptor structure for the carbohydrate derivatization and simultaneously isolate the electrode layer from the interaction interface. A range of polymers were initially envisaged, including poly(2-ethyl-2-oxazoline) (PEOX), poly(ethylene glycol) (PEG), and poly(ethylene oxide) (PEO). Of these, the PEG- and PEO-type polymers were chosen for the present application due to their reputed biocompatibility, known to prevent nonspecific protein and cell adhesion. However, these polymers cannot be permanently physisorbed to the gold electrode, in contrast to, for example, polystyrene,32 for which reason a covalent surface ligation strategy was developed. PFPA-Chemistry was in this case preferably chosen, thus leading to a double surface ligation methodology in combination with the carbohydrate derivatization.
The perfluorophenylazide-based double surface ligation method is outlined in Figure 1. The gold surfaces of the QCM crystals were first covalently activated to establish a PFPA layer. This was achieved by allowing a PFPA–disulfide derivative (compound 3) to bind to the gold surface using conventional disulfide to gold coupling. This PFPA-activated surface was subsequently used to covalently attach a thin layer of PEG. Both photolytic and thermolytic nitrene generation could in this cases be used to attach the polymer, where thermoinitiation proved favorable for the finally chosen polymer due to the higher ease of operation. The combined action of all inserted PFPA-structures ensured a strong attachment of the polymer to the gold surface. In order to evaluate the surface behavior, different PEG- or PEO-structures were also tested, ranging in molecular weight from 6000 to 200 000 g/mol. Shorter polymers resulted in increased nonspecific binding, whereas polymers of higher molecular weight proved somewhat sensitive to the flow-through system. High molecular weight PEO (Mr 200 000) showed very low nonspecific binding but did not perform optimally in the flow-through system, where repeated injections of lectin resulted in gradually diminished binding. These effects are currently under more detailed investigation. The medium molecular weight PEG (Mr 20 000) was in this case found to possess the best overall performance, resulting in a very low level of nonspecific binding and, at the same time, high durability of the surfaces.
After rinsing of the crystals, a thin monolayer of PEG remained on the surface,5 efficiently covering the underlying gold layer and producing a highly hydrophilic, biocompatible surface. The resulting polymer layer then served as a substrate for photoligation of different carbohydrate structures. Three different PFPA-derivatized carbohydrates were used as prototype surface recognition motifs: α-d-mannopyranoside 4, β-d-galactopyranoside 5, and α-l-fucopyranoside 6 (Figure 1). The parent monosaccharides represent building blocks of natural glycans and are known targets for specific carbohydrate-binding proteins. This enabled direct estimation of the lectin binding efficiency, where crossreactivities could be easily monitored. Derivatization of the structures at the anomeric position was expected to lead to nonsignificant selectivity effects and could even improve affinity. Initial attempts with reduced distance between the PFPA moiety and the carbohydrate group showed that the length and nature of the linker was, as expected, important for efficient interactions. Thus, in order to ensure efficient protein binding to the surfaces, a sufficiently long ethylene glycol-based linker spacer had to be inserted, yielding a 16-bond distance between the anomeric oxygen and the polymer surface (~18 Å in the extended state). Application of these carbohydrate-derivatized photoprobe structures to the surfaces, and exposure by short-time UV-irradiation, resulted in covalently conjugated carbohydrate derivatives to the polymer layer.
The resulting carbohydrate-functionalized QCM crystals were subsequently subjected to analysis of bioactivity with respect to carbohydrate–protein interactions. The crystals were mounted in the flow-through QCM system and equilibrated with running buffer solution (10 mM PBS, pH 7.4) at a flow rate of 50 µL/min until no frequency change was recorded. The carbohydrate-binding proteins, dissolved in the running buffer, were subsequently injected (50 µL), and binding was monitored as reduced oscillation frequency of the quartz crystal. After each step of analysis, the bound lectins were removed by two successive injections of low pH buffer (pH 1.5) to regenerate the surface without diminishing the performance of the sensor response.
Protein binding to the carbohydrate-presenting surfaces was evaluated for a set of four different plant lectins:36,37 the agglutinin from Canavalia ensiformis (Con A), Viscum album agglutinin (VAA), Ulex europaeus lectin I (UEA), and Pisum sativum agglutinin (PSA). These lectins were chosen due to their known carbohydrate specificities, compatible with the parent carbohydrate structures. PSA and Con A are both specific for α-d-mannopyranosides, albeit with different binding strengths, UEA specifically binds α-l-fucopyranoside structures, and VAA binds to β-d-galactopyranosides.
Figure 2 displays typical frequency-response graphs recorded for Con A-binding to the three different carbohydrate-derivatized surfaces. On the basis of the carbohydrate specificity, only the α-d-mannopyranoside surface should serve as a sensor recognition element for Con A, whereas the other surfaces should be inefficient. As can be seen, the α-d-mannopyranoside surface indeed proved highly efficient in binding this lectin, resulting in a large maximum response. On the other hand, the β-d-galactopyranoside- and the α-l-fucopyranoside-derivatized surfaces did not show any residual binding to this lectin, indicating that the interaction is governed by carbohydrate–protein recognition. Apparent transient binding to both surfaces could however be recorded, where especially the α-l-fucopyranoside-surface showed temporary binding of the lectin to approximately 60% of the maximal level of binding to the α-d-mannopyranoside-derivatized surface upon the initial injection of Con A. As the concentration of Con A in the flow cell was diluted and eventually replaced by the running buffer, the weakly bound Con A was removed from the surface. Binding to the β-d-galactopyranoside-presenting surface represents in this case a more standard behavior of a nonspecific surface, where a low amount of adsorption to the hydrophilic surface takes place during the protein flow over the crystal. Thus, the system can potentially be used to monitor and quantitate weaker, transient binding, as well as stronger affinity. These effects are currently under more thorough investigations.
Figure 2.

Comparison of the binding of Con A to different carbohydrates immobilized on the PEG-coated surface. Surfaces: α-d-mannopyranoside (Man), β-d-galactopyranoside (Gal), and α-l-fucopyranoside (Fuc).
The overall binding results for the four different plant lectins to all three carbohydrate-presenting surfaces are displayed in Figure 3. In order to compare the results between the lectins, the frequency responses were related to the molecular weight (Mr) of the respective lectin (Mr: Con A 104 000;38,39 PSA 48 000;40 VAA 119 800;41 UEA 63 00042). The resulting Δf/Mr-values represent the relative binding of protein to the surface, irrespective of the differences in mass. Since Δf corresponds to the applied mass to the surface, subject to prerequisites of the Sauerbrey equation,43 the Δf/Mr-value corresponds to the amount of substance of protein. The results reflect the expected binding patterns for these lectins, where the α-d-mannopyranoside-presenting surface was selective for PSA and Con A, the β-d-galactopyranoside-presenting surface was selective for VAA, and the α-l-fucopyranoside-presenting surface selective for the lectin from Ulex europaeus (UEA). In either case, no significant cross-reactivity between lectins of different specificity could be detected, in agreement with data from solid-phase- and histochemical assays based on carbohydrate-presenting natural and artificial surfaces.36,44–46 At the present concentrations, the binding performance for three of the lectins (VAA, Con A, and UEA) showed similar specific responses to the respective surfaces (Δf = 0.7–1 mHz/Mr), indicative of near surface saturation. On the other hand, the PSA-lectin showed a lower specific response (Δf = 0.3 mHz/Mr) compared to Con A, indicating that PSA possesses lower binding capability to the α-d-mannopyranoside surface. This effect is also in accordance with literature data, where PSA shows lower binding affinity to α-d-mannopyranosides than the Con A lectin.47,48
Figure 3.

Determination of binding selectivity. Residual binding (columns) for all four different lectins (Con A, PSA, VAA, UEA) recorded for all three different carbohydrate-derivatized surfaces: α-d-mannopyranoside (Man), β-d-galactopyranoside (Gal), and α-l-fucopyranoside (Fuc). A horizontal bar indicates no detectable residual binding. Protein concentration 10 µM (Con A, PSA, UEA) or 5 µM (VAA).
The binding characteristics of Con A to the α-d-mannopyranoside-presenting surface were subsequently evaluated by saturation analysis in order to further estimate the utility of the methodology developed. Samples of Con A ranging in concentration from 5 nM to 10 µM were injected over the α-d-mannopyranoside-presenting surface and the frequency differences resulting from the Con A-binding recorded. After each sample injection, the surface was regenerated by two successive injections of low pH buffer. The resulting saturation isotherm is displayed in Figure 4, indicating that the surface is completely saturatable and the binding follows Langmuir-like isothermal behavior. The apparent dissociation constant could be estimated to KD = 0.54 µM (95% CI: 0.44–0.64 µM), a value well in accordance with reported literature values (0.1–1.1 µM).32,49,50 These results show that the technique can be used also to estimate affinity constants between the surfaces developed and a protein binding partner.
Figure 4.

Saturation binding of Con A to the α-d-mannopyranoside-presenting surface. KD = 0.54 µM (95% CI: 0.44–0.64 µM).
CONCLUSIONS
In conclusion, it has been shown that efficient bioactive surfaces can be prepared by a double surface ligation methodology based on perfluorophenylazide chemistry. Gold surfaces, functionalized with azide groups, could be covalently coated with thin layers of poly(ethylene glycol), to which photoprobe-derivatized carbohydrate structures could be covalently attached by a nitrene photoinsertion reaction. The resulting surfaces could be applied to a flow-through QCM instrument, providing a robust, biocompatible system enabling analysis of carbohydrate–protein interactions in real time. This was demonstrated for a series of plant lectins, evaluated for their binding to different carbohydrate-presenting surfaces, where specificity and binding strength could be efficiently estimated. The technique is however not limited to carbohydrate–protein interactions but provides a versatile means to a wide range of different affinity systems. The developed photoprobe can be conjugated to a variety of small molecules, as well as macromolecular binding partners, and directly applied to the polymer surfaces. Thus, the technique opens a future scope to sensing of a plethora of recognition systems. Given more in-depth understanding of transient binding, currently under exploration, the technique can furthermore provide kinetic and thermo-dynamic data for the interactions studied.
ACKNOWLEDGMENT
This work was supported by the Wenner-Gren foundation, NIH AREA Award 1R15 GM066279-01A2, the European Commission Contract MRTN-CT-2005-019561, the Swedish Research Council, the Royal Institute of Technology, and the travel exchange program between the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) and the Deutscher Akademischer Austauschdienst (DAAD).
References
- 1.Scriven EFU. Azides and Nitrenes: Reactivity and Utility. New York: Academic Press; 1984. [Google Scholar]
- 2.Keana JFW, Cai SX. J. Fluorine Chem. 1989;43:151. [Google Scholar]
- 3.Banks RE, Sparkes GR. J. Chem. Soc., Perkin Trans. 1972;1:2964. [Google Scholar]
- 4.Keana JFW, Cai SX. J. Org. Chem. 1990;55:3640. [Google Scholar]
- 5.Bartlett MA, Yan M. Adv. Mater. 2001;13:1449. [Google Scholar]
- 6.Liu L, Yan M. Angew. Chem., Int. Ed. 2006;45:6207. doi: 10.1002/anie.200602097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu L, Engelhard MH, Yan M. J. Am. Chem. Soc. 2006;128:14067. doi: 10.1021/ja062802l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan M, Bartlett M. Nano Lett. 2002;2:275. [Google Scholar]
- 9.Yan M, Ren J. Chem. Mater. 2004;16:1627. [Google Scholar]
- 10.Yarema KJ, Bertozzi CR. Curr. Opin. Chem. Biol. 1998;2:49. doi: 10.1016/s1367-5931(98)80035-5. [DOI] [PubMed] [Google Scholar]
- 11.Wong C-H. Acc. Chem. Res. 1999;32:376. [Google Scholar]
- 12.Bertozzi CR, Kiessling LL. Science. 2001;291:2357. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 13.Gabius H-J, Siebert H-C, André S, Jiménez-Barbero J, Rüdiger H. Chem Bio Chem. 2004;5:740. doi: 10.1002/cbic.200300753. [DOI] [PubMed] [Google Scholar]
- 14.André S, Maljaars CEP, Halkes KM, Gabius H-J, Kamerling JP. Bioorg. Med. Chem. Lett. 2007;17:793. doi: 10.1016/j.bmcl.2006.10.067. [DOI] [PubMed] [Google Scholar]
- 15.Pei Z, Yu H, Theurer M, Waldén A, Nilsson P, Yan M, Ramström O. Chem Bio Chem. 2007;8:166. doi: 10.1002/cbic.200600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duverger E, Frison N, Roche A-C, Monsigny M. Biochimie. 2003;85:167. doi: 10.1016/s0300-9084(03)00060-9. [DOI] [PubMed] [Google Scholar]
- 17.Holmskov U, Fischer PB, Rothmann A, Hojrup P. FEBS Lett. 1996;393:314. doi: 10.1016/0014-5793(96)00915-5. [DOI] [PubMed] [Google Scholar]
- 18.Pagé D, Zanini D, Roy R. Bioorg. Med. Chem. 1996;4:1949. doi: 10.1016/s0968-0896(96)00177-0. [DOI] [PubMed] [Google Scholar]
- 19.Ebara Y, Okahata Y. J. Am. Chem. Soc. 1994;116:11209. [Google Scholar]
- 20.Meyer B, Peters T. Angew. Chem., Int. Ed. 2003;42:864. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 21.Paulson JC, Blixt O, Collins BE. Nat. Chem. Biol. 2006;2:238. doi: 10.1038/nchembio785. [DOI] [PubMed] [Google Scholar]
- 22.Smith EA, Thomas WD, Kiessling LL, Corn RM. J. Am. Chem. Soc. 2003;125:6140. doi: 10.1021/ja034165u. [DOI] [PubMed] [Google Scholar]
- 23.Dam TK, Brewer CF. Chem. Rev. 2002;102:387. doi: 10.1021/cr000401x. [DOI] [PubMed] [Google Scholar]
- 24.Janshoff A, Galla HJ, Steinem C. Angew. Chem., Int. Ed. 2000;39:4004. doi: 10.1002/1521-3773(20001117)39:22<4004::aid-anie4004>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 25.O’Sullivan CK, Guilbault GG. Biosens. Bioelectron. 1999;14:663. [Google Scholar]
- 26.Miura Y, Sasao Y, Dohi H, Nishida Y, Kobayashi K. Anal. Biochem. 2002;310:27. doi: 10.1016/s0003-2697(02)00318-4. [DOI] [PubMed] [Google Scholar]
- 27.Hildebrand A, Schaedlich A, Rothe U, Neubert RHH. J. Colloid Interface Sci. 2002;249:274. doi: 10.1006/jcis.2002.8272. [DOI] [PubMed] [Google Scholar]
- 28.Bakowsky U, Rettig W, Bendas G, Vogel J, Bakowsky H, Harnagea C, Rothe U. Phys. Chem. Chem. Phys. 2000;2:4609. [Google Scholar]
- 29.Hashizume M, Sato T, Okahata Y. Chem. Lett. 1998:399. doi: 10.1023/a:1009273827598. [DOI] [PubMed] [Google Scholar]
- 30.Steinem C, Janshoff A, Wegener J, Ulrich W-P, Willenbrink W, Sieber M, Galla H-J. Biosens. Bioelectron. 1997;12:787. doi: 10.1016/s0956-5663(97)00045-6. [DOI] [PubMed] [Google Scholar]
- 31.Pei Z, Aastrup T, Anderson H, Ramström O. Bioorg. Med. Chem. Lett. 2005;15:2707. doi: 10.1016/j.bmcl.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 32.Pei Z, Anderson H, Aastrup T, Ramström O. Biosens. Bioelectron. 2005;21:60. doi: 10.1016/j.bios.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Pei Z, Larsson R, Aastrup T, Anderson H, Lehn J-M, Ramström O. Biosens. Bioelectron. 2006;22:42. doi: 10.1016/j.bios.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 34.Pyykkö P. Angew. Chem., Int. Ed. 2004;43:4412. doi: 10.1002/anie.200300624. [DOI] [PubMed] [Google Scholar]
- 35.Jimenez M, Andre S, Siebert H-C, Gabius H-J, Solís D. Glycobiology. 2006;16:926. doi: 10.1093/glycob/cwl017. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein IJ, Poretz RD. In: The Lectins. Properties, Functions, and Applications in Biology and Medicine. Liener IE, Sharon N, Goldstein IJ, editors. Orlando, FL: Academic Press; 1986. p. 33. [Google Scholar]
- 37.Rüdiger H, Gabius H-J. Glycoconjugate J. 2001;18:589. doi: 10.1023/a:1020687518999. [DOI] [PubMed] [Google Scholar]
- 38.McKenzie GH, Sawyer WH, Nichol LW. Biochim. Biophys. Acta. 1972;263:283. doi: 10.1016/0005-2795(72)90081-5. [DOI] [PubMed] [Google Scholar]
- 39.Wang JL, Cunningham BA, Waxdal MJ, Edelman GM. J. Biol. Chem. 1975;250:1490. [PubMed] [Google Scholar]
- 40.Trowbridge IS. J. Biol. Chem. 1974;249:6004. [PubMed] [Google Scholar]
- 41.Jiménez M, Sáiz JL, André S, Gabius H-J, Solís D. Glycobiology. 2005;15:1386. doi: 10.1093/glycob/cwj020. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Kochetkova I, Haddad A, Hoyt T, Hone DM, Pascual DW. Vaccine. 2005;23:3836. doi: 10.1016/j.vaccine.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 43.Sauerbrey G. Z. Phys. 1959;155:206. [Google Scholar]
- 44.Lee RT, Gabius H-J, Lee YC. J. Biol. Chem. 1992;267:23722. [PubMed] [Google Scholar]
- 45.Galanina OE, Kaltner H, Khraltsova LS, Bovin NV, Gabius H-J. J. Mol. Recognit. 1997;10:139. doi: 10.1002/(SICI)1099-1352(199705/06)10:3<139::AID-JMR358>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 46.Manning JC, Seyrek K, Kaltner H, André S, Sinowatz F, Gabius H-J. Histol. Histopathol. 2004;19:1043. doi: 10.14670/HH-19.1043. [DOI] [PubMed] [Google Scholar]
- 47.Schwarz FP, Puri KD, Bhat RG, Surolia A. J. Biol. Chem. 1993;268:7668. [PubMed] [Google Scholar]
- 48.Schlick KH, Udelhoven RA, Strohmeyer GC, Cloninger MJ. Mol. Pharm. 2005;2:295. doi: 10.1021/mp050014h. [DOI] [PubMed] [Google Scholar]
- 49.Mislovicova D, Masarova J, Svitel J, Mendichi R, Soltes L, Gemeiner P, Danielsson B. Bioconjugate Chem. 2002;13:136. doi: 10.1021/bc015517u. [DOI] [PubMed] [Google Scholar]
- 50.Horisberger M. Biochim. Biophys. Acta. 1980;632:298. doi: 10.1016/0304-4165(80)90088-4. [DOI] [PubMed] [Google Scholar]