

Summary
The mechanisms by which the microtubule cytoskeleton regulates the permeability of endothelial barrier are not well understood. Here, we demonstrate that microtubule-associated end-binding protein 3 (EB3), a core component of the microtubule plus-end protein complex, binds to inositol 1,4,5-trisphosphate receptors (IP3Rs) through an S/TxIP EB-binding motif. In endothelial cells, α-thrombin, a pro-inflammatory mediator that stimulates phospholipase Cβ, increases the cytosolic Ca2+ concentration and elicits clustering of IP3R3s. These responses, and the resulting Ca2+-dependent phosphorylation of myosin light chain, are prevented by depletion of either EB3 or mutation of the TxIP motif of IP3R3 responsible for mediating its binding to EB3. We also show that selective EB3 gene deletion in endothelial cells of mice abrogates α-thrombin-induced increase in endothelial permeability. We conclude that the EB3-mediated interaction of IP3Rs with microtubules controls the assembly of IP3Rs into effective Ca2+ signaling clusters, which thereby regulate microtubule-dependent endothelial permeability.
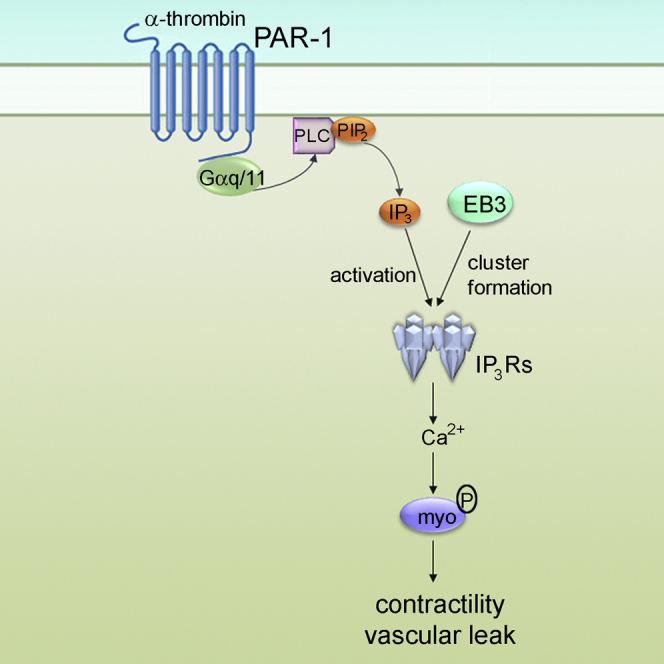
Graphical Abstract
Highlights
-
•
IP3 receptors (IP3Rs) bind to microtubule end-binding protein EB3
-
•
EB3, in turn, promotes IP3R clustering and Ca2+ signals in endothelial cells
-
•
IP3R-EB3 interaction thereby contributes to endothelial barrier disruption
-
•
In vivo EB3 deletion in endothelial cells protects from vascular hyperpermeability
End binding proteins (EBs) mediate interactions between growing microtubules and intracellular structures. Geyer et al. demonstrate that interactions between EB3 and IP3 receptors control clustering of IP3 receptors in endothelial cells and Ca2+ signaling, and thus permeability of endothelial barrier in inflammatory diseases.
Introduction
Adherens junctions (AJs) responsible for endothelial cell interactions (Dejana, 2004) and acto-myosin contraction (Wainwright et al., 2003) regulate the integrity of the vascular endothelial barrier. The ectodomain of vascular endothelial (VE)-cadherin, which is the primary adhesion molecule of AJs, undergoes homophilic trans-dimerization to form AJs, while its intracellular domain interacts with the actin cytoskeleton (Daneshjou et al., 2015, Giannotta et al., 2013). Activation of phospholipase C (PLC) via G protein-coupled receptors (GPCRs), such as the protease-activated receptor-1 (PAR-1), or by disruption of VE-cadherin trans-interactions (Komarova et al., 2012), causes increased endothelial permeability (Komarova and Malik, 2010). An increase in cytosolic Ca2+ concentration ([Ca2+]c), by promoting disassembly of AJs and acto-myosin-mediated contraction of endothelial cells, is a crucial signal mediating this increased endothelial permeability (Komarova and Malik, 2010).
The roles of actin polymerization and disassembly of AJs in increasing endothelial permeability have received considerable attention, but microtubules may also contribute to the response by poorly understood mechanisms (Vogel and Malik, 2012). Microtubules are polarized tubular filaments of heterodimers of α- and β-tubulin, with distinct plus and minus ends (Howard and Hyman, 2003). Plus ends, usually directed toward the cell periphery, undergo cycles of polymerization and depolymerization regulated by plus-end tracking proteins (+TIPs) (Akhmanova and Steinmetz, 2010). The end-binding proteins, EB1 and EB3, members of the RP/EB family, are core elements of the dynamic +TIP complex. EBs transiently bind to growing microtubules by recognizing the GTP-bound state of β-tubulin (Maurer et al., 2012). EB binding enhances lateral contacts between tubulin molecules and prevents the transition from microtubule growth to shrinkage (“catastrophe” events) (Komarova et al., 2009, Maurer et al., 2012). In addition, EBs provide an essential hub for assembly of other +TIPs that facilitate interactions of microtubules with various macromolecules and organelles (Akhmanova and Steinmetz, 2010). The latter include endoplasmic reticulum (ER), which is continuously remodeled through its interactions with microtubules (Pendin et al., 2011). These interactions involve both tethering of the ER protein, stromal interaction molecule 1 (STIM1), to the +TIP complex by EB1, and the association of ER tubules with the plus-end directed microtubule motor protein, kinesin 1 (Friedman and Voeltz, 2011, Grigoriev et al., 2008).
ER is the major intracellular Ca2+ store. Inositol 1,4,5-trisphosphate receptors (IP3Rs) within ER membranes allow rapid release of Ca2+ from the ER (Foskett et al., 2007, Taylor et al., 2014). Emptying of ER Ca2+ stores then causes clustering of STIM1, and this activates store-operated Ca2+ entry into the cell across the plasma membrane (Wu et al., 2014). The release of Ca2+ from the ER evoked by IP3 proceeds through recruitment of Ca2+ release events that depend on IP3 priming IP3Rs to respond to Ca2+. This form of regulation allows clustered IP3Rs to stimulate the activity of their neighbors by Ca2+-induced Ca2+ release (Smith et al., 2009). The lowest concentrations of IP3 stimulate openings of single IP3Rs, and as the IP3 concentration increases the Ca2+ released by active IP3Rs is thought to stimulate coordinated opening of IP3Rs within a cluster, generating a Ca2+ “puff.” Further increases in IP3 concentration ignite regenerative Ca2+ waves that spread across the cell (Smith et al., 2009). The recruitment of IP3R activity depends critically on the distribution of IP3Rs in ER membranes, wherein most IP3Rs appear to be mobile (Ferreri-Jacobia et al., 2005, Pantazaka and Taylor, 2011). Stimulation of PLC causes reversible clustering of IP3Rs in cells (Chalmers et al., 2006, Tateishi et al., 2005) and within nuclear envelope IP3 causes IP3Rs to cluster (Taufiq-Ur-Rahman et al., 2009). Importantly in the context of the present study, IP3Rs associate with microtubules (Takei et al., 1998) and the association contributes to the redistribution of IP3Rs during sustained stimulation (Vermassen et al., 2003), extension of neuronal growth cones (Zhang and Forscher, 2009), and cell division (Mitsuyama and Sawai, 2001). There is, however, also evidence that IP3R clustering can persist after disruption of microtubules (Wilson et al., 1998, Taufiq-Ur-Rahman et al., 2009).
Studies using drugs (colchicine, nocodazole, and taxol) suppressing microtubule dynamics suggest an important role of microtubule cytoskeleton in organizing IP3-evoked Ca2+ signals (Isshiki et al., 1998, Fogarty et al., 2000). Perturbing the microtubule cytoskeleton inhibits receptor-activated release of Ca2+ via IP3Rs (Tasaka et al., 1991), prevents initiation of Ca2+ waves (Béliveau and Guillemette, 2009, Isshiki et al., 1998), slows diffusion of IP3Rs within ER membranes (Ferreri-Jacobia et al., 2005), disrupts local delivery of IP3 to IP3Rs (Graier et al., 1998, Ribeiro et al., 1997), and abolishes IP3-activated Ca2+ spikes at the apical pole of secretory cells (Fogarty et al., 2000). Many of these perturbations might result from effects of microtubules in organizing the ER, but there is also the possibility of more direct interactions with IP3Rs. The latter would be significant for vascular endothelial cells, where we have shown that PLC-evoked Ca2+ signals cause dephosphorylation of EB3 leading to persistent growth of microtubules, disassembly of AJs, and increased endothelial permeability (Komarova et al., 2012).
Here, we demonstrate a direct interaction between EB3 and the S/TxIP motif within IP3R3 that allows IP3Rs to associate with growing microtubule tips. Depletion of EB3 or disruption of the interaction with an IP3R3 point mutation prevents both clustering of IP3R3s and Ca2+ signals elicited by activation of PAR-1. Further, selective deletion of the EB3 gene in endothelium in mice inhibits the increase in endothelial permeability elicited by activation of PAR-1. Thus, microtubule-associated EB3 plays an obligatory role in organizing IP3-induced Ca2+ signaling, and, in turn, regulating endothelial permeability.
Results
Loss of EB3 Impairs Ca2+ Signaling in Endothelial Cells
In primary cultures of human lung microvascular endothelial cells (HLMVECs), α-thrombin stimulates PAR-1, a GPCR that causes activation of PLCβ, formation of IP3 and release of Ca2+ from intracellular stores. Depletion of EB3, using small interfering RNA (siRNA) (Figure S1A), significantly reduced the amplitude of the Ca2+ signals evoked by addition of α-thrombin in Ca2+-free medium and the subsequent response to restoration of extracellular Ca2+ (Figures 1A and 1B). The inhibition was substantially reversed by expression of a siRNA-resistant EB3-GFP, but not by EB1-GFP. Loss of EB1 had no significant effect on α-thrombin-activated Ca2+ signals. A C-terminal fragment of EB3 (EB3-Ct-mRFP), which prevents binding of endogenous EBs to microtubule tips by forming non-functional dimers with endogenous proteins (Komarova et al., 2009), also inhibited α-thrombin-induced Ca2+ signals (Figures S1B and S1C).
Figure 1.

Depletion of EB3 Attenuates Receptor-Induced Ca2+ Release from ER
(A) Cytosolic Ca2+ signals evoked by addition of α-thrombin (50 nM) recorded from ∼5–10 fura-2-loaded HLMVECs in Ca2+-free medium, followed by restoration of extracellular Ca2+ (2 mM). Cells were treated with siRNA for EB1 or EB3, control siRNA, or siRNA for EB3 together with expression of EB1-GFP or a siRNA-resistant EB3-GFP.
(B) Summary results show peak changes in [Ca2+]c (Δ[Ca2+]c) evoked by α-thrombin (Ca2+ release) and the subsequent restoration of extracellular Ca2+ (Ca2+ entry). Results show data points color coded as in (A), and mean ± SEM from four to eight experiments with five to ten cells analyzed in each group. ∗,∗∗,∗∗∗p values are relative to control siRNA-treated cells using one-way ANOVA.
(C) Time-lapse images (times shown in seconds) show overlaid red (mCherry-ER) and green (G-CEPIA1er) fluorescence for cells treated with control or EB3 siRNA. Cells were stimulated with α-thrombin (50 nM at t = 0) in Ca2+-free medium, before restoration of extracellular Ca2+. Loss of luminal Ca2+ causes the green fluorescence of the luminal Ca2+ indicator to decrease, shifting images from yellow to red. The first panel shows an enlargement of the boxed area. Scale bars, 1 (enlargement) and 10 μm (other panels).
(D) Experiments similar to those in (C) show the time course of the changes in [Ca2+]ER presented as fluorescence ratios (G-CEPIA/mCherry-ER) normalized to the ratio recorded 4 s before addition of α-thrombin (100%) for cells treated with control or EB3 siRNA.
(E) Summary results (mean ± SEM from 6 to 16 cells) show [Ca2+]ER measured before (basal) and after stimulation with α-thrombin (50 nM) using GEM-CEPIA1er in cells treated with control or EB3 siRNA. Results are from two independent experiments; ∗∗∗relative to matched siRNA treatment without α-thrombin stimulation, using one-way ANOVA.
We used low-affinity, genetically encoded Ca2+ indicators expressed in the lumen of the ER, G-CEPIA1er and the ratiometric indicator GEM-CEPIA1er (Suzuki et al., 2014), to establish whether loss of EB3 affected the Ca2+ content of the intracellular stores. Depletion of EB3 affected neither the organization of the ER nor the free Ca2+ concentration within the ER ([Ca2+]ER) (Figures 1C–1E). [Ca2+]ER was 583 ± 77 μM and 489 ± 53 μM (n = 6–16 cells) in control and EB3 siRNA-treated cells, respectively (Figure 1E). Stimulation of control cells with α-thrombin caused [Ca2+]ER to fall to 96 ± 33 μM, whereas α-thrombin had no significant effect on cells treated with siRNA for EB3 (436 ± 122 μM) (Figure 1E). Furthermore, refilling of ER after restoration of extracellular Ca2+ to cells stimulated with α-thrombin in Ca2+-free medium was faster in cells lacking EB3 (Figure 1D), consistent with reduced activation of IP3Rs after knockdown of EB3. These results demonstrate that loss of EB3 attenuates the release of Ca2+ from intracellular stores elicited by α-thrombin without affecting the initial Ca2+ content of the ER.
EB3 Binds to IP3Rs
To assess whether the effects of EB3 on Ca2+ release involved reorganization of the microtubule network, we analyzed changes in microtubule dynamics after addition of α-thrombin using time-lapse confocal imaging of EB1-GFP to mark growing microtubule tips. We chose EB1-GFP for this analysis because it does not rescue the inhibition of α-thrombin-evoked Ca2+ signals after EB3 depletion, and nor does loss of endogenous EB1 affect Ca2+ signals (Figures 1A and 1B). In confluent monolayers of HLMVECs treated with control siRNA, microtubules grew at 13.7 ± 3.1 μm/min, and they displayed frequent catastrophe events. Depletion of EB3 had no effect on the growth rate or catastrophe frequency (Table 1), suggesting that EB3 did not affect microtubule dynamics under basal conditions. This finding is consistent with previous work demonstrating that most EB3 is phosphorylated in unstimulated HLMVECs, and therefore unable to promote persistent growth of microtubules (Komarova et al., 2012). In the 2–3 min after stimulation of PAR-1 with α-thrombin, there was no effect on the catastrophe frequency, but the stimulation unexpectedly reduced microtubule growth rate in both control and EB3-siRNA-treated cells (Table 1; Figure S2). These results demonstrate that loss of EB3 has no discernible effect on microtubule dynamics under conditions where it attenuates α-thrombin-evoked Ca2+ release. We therefore considered whether EB3 might regulate PLC or IP3Rs.
Table 1.
Effects of α-Thrombin and Loss of EB3 on Microtubule Dynamics
| Growth Rate (μm/min) | Catastrophe Frequency (min–1) | |
|---|---|---|
| Basal | ||
| Control siRNA | 13.7 ± 3.1 | 6.2 ± 4.2 |
| EB3 siRNA | 14.1 ± 2.9 | 5.5 ± 2.2 |
| α-thrombin | ||
| Control siRNA | 11.0 ± 3.0a | 7.4 ± 4.6 |
| EB3 siRNA | 12.1 ± 3.0a | 6.6 ± 6.5 |
Growth rates of microtubules were calculated from the histogram of instantaneous displacement rates of microtubule tips between frames collected every 3 s (Figure S2). Catastrophe frequency was calculated from the number of shortening events per min (Supplemental Experimental Procedures). Results are means ± SD (n = 7–8 cells).
Paired two-tailed Student’s t test compared to unstimulated cells.
We used the IP3 biosensor, LIBRAvIII, in which IP3 binding causes a decrease in intramolecular fluorescence resonance energy transfer (FRET) (Tanimura et al., 2009), to measure cytosolic IP3 concentrations in confluent monolayers of HLMVECs. α-Thrombin caused a decrease in the FRET signal consistent with an increase in cytosolic IP3 concentration. The response was similar in cells treated with control siRNA or siRNA to EB3 (Figure S3A). These results thus suggest that under conditions where loss of EB3 attenuates α-thrombin-evoked Ca2+ signals, EB3 has no effect on α-thrombin-induced IP3 formation.
Alignment of IP3R sequences identified a conserved SxIP motif within a short region of disordered protein structure in all three mammalian IP3R subtypes (Figure S3B). This motif (residues 804–807 in human IP3R3), a signature of EB-interacting partners (Honnappa et al., 2009), is located downstream of the IP3-binding site. The interaction between full-length EB3 or its C-terminal region (residues 200–281), and IP3R3 was demonstrated in pull-down assays using immobilized (His6)-EBs expressed in bacteria and GFP-IP3Rs from lysates of HEK cells (Figures 2A, 2B, and S3C). IP3R1 and IP3R2 also associated with full-length EB3 (Figure 2C). EB1 was less effective than EB3 in the pull-downs of GFP-IP3R3 (Figure 2B). For each IP3R subtype, the interaction with EB3 was weaker than that between EB3 and STIM1, another Ca2+ signaling protein to which EB1 and EB3 bind (Grigoriev et al., 2008) (Figure S3D). Interaction between endogenous EB3 and IP3R3 was confirmed by their co-immunoprecipitation (Figure S3E). Deletion of the acidic C-terminal tail of EB3 (EB3ΔAc), which contributes to the binding interface for the S/TxIP motif, abolished the interaction of IP3R3 with EB3 (Figure 2B). Mutation of the critical Thr within the TxIP motif of IP3R3 (T804A) also abolished the interaction with EB3 (Figure 2C). These results suggest a direct interaction between the C-terminal region of EB3 and the TxIP motif of IP3R3, an interaction that is probably shared with other IP3R subtypes.
Figure 2.

EB3 Interacts Directly with IP3Rs
(A) Schematic representation of hexa-histidine (His6)-tagged EB constructs used for pull-down experiments. CHD, calponin homology domain; L, linker; CC, coiled coil; Ac, acidic tail.
(B and C) Pull-down analyses of interactions between (His6)-EB proteins covalently bound to Ni-NTA resin and lysates from HEK cells expressing GFP-IP3R1-3 or GFP-IP3R3(T804A). Upper panels show western blots for GFP and lower panels show Coomassie brilliant blue-stained gels loaded with 5% of the EBs (numbered as in A) used for the pull-down. Results are typical of three independent experiments. Western blots of GFP-IP3Rs in the cell lysates used and additional controls are shown in Figure S3.
(D) Confocal images collected at 850-ms intervals show simultaneous recordings of fluorescence from EB3-mRFP and GFP-IP3R3 in HLMVECs. The composite panels show overlaid GFP-IP3R3 (green) and EB3-mRFP (red). Note the loss of GFP-IP3R3 fluorescence (circle) as the EB3-mRFP-labeled microtubule tip approaches (arrow). Scale bar, 5 μm.
(E) EB3-mRFP and GFP-IP3R3 fluorescence recorded along the dashed line shown in the merged images in (D) illustrates the decrease in GFP fluorescence at the point of interaction with the microtubule tip.
(F) Focal photobleaching of the acceptor fluorophore (mRFP) at the microtubule tip while recording donor fluorescence from GFP-IP3R was used to assess the interaction between GFP-IP3R3 and EB3-mRFP or CLIP-170-mRFP at the microtubule tip. Individual data points with mean ± SEM from five to eight cells analyzed in each group show the recovery of the donor fluorescence after acceptor photobleaching (%). ∗∗Using Student’s t test.
In HLMVECs expressing EB3-mRFP and GFP-IP3R3, there were transient contacts between growing microtubule tips and GFP-IP3R3 in ER membranes (Figure 2D). However, GFP-IP3R3 did not form the “comet-like” structures described for STIM-1 associated with growing microtubule tips (Grigoriev et al., 2008, Pozo-Guisado et al., 2010). This finding is consistent with the lower affinity of EB3 relative to STIM1 for IP3R3, because comets reveal the density of EB proteins, which declines with distance from the microtubule tip. We also observed quenching of the GFP fluorescence when EB3-mRFP-labeled microtubule tips made contact with ER tubules, suggesting an intermolecular FRET between EB3-mRFP and GFP-IP3R3 (Figures 2D and 2E). Focal photobleaching of the EB3-mRFP acceptor caused a transient increase in closely apposed fluorescence of the GFP-IP3R3 donor (Figure 2F), confirming an interaction between EB3 and IP3R3 in intact cells. There was no detectable FRET between GFP-IP3R3 and another +TIP, mRFP-CLIP-170 (Figure 2F). The results thus indicate a specific interaction between EB3 and IP3R3 in intact human lung endothelial cells.
Interactions between EB3 and IP3R3 Regulate IP3R3 Dynamic and Activity
Analysis of mRNA and protein expression demonstrated that in various human pulmonary endothelial cells, including HLMVECs, IP3R2 and IP3R3 were the major subtypes (Figures S4A–S4C). Immunostaining of HLMVECs revealed that IP3R3 formed puncta (Figure 3A), consistent with their assembly into clusters, as reported for IP3Rs in endothelial cells (Tran et al., 2014). Depletion of EB3 had no effect on the expression of IP3R3 (Figure S4D), but it significantly reduced the number of IP3R3 clusters (Figures 3A and 3B). EB3 depletion had no effect on IP3R2 clusters, which were observed in the perinuclear region in both control and EB3 siRNA-treated cells (Figures S4E and S4F). The inhibition of IP3R3 clustering by depletion of EB3 was reversed by expression of a siRNA-resistant EB3, but not by expression of the EB3ΔAc mutant (Figure 3B) that does not bind to IP3R3 (Figure 2B).
Figure 3.

EB3 Facilitates Clustering of IP3R3
(A) Intracellular distribution of endogenous IP3R3 (immunostaining) in HLMVECs treated with control or EB3 siRNA. Central panels show enlargements of the boxed area. The thresholded images used to measure cluster densities (see Experimental Procedures) are shown in the right panels. Scale bar, 10 μm.
(B) Summary results from nine to 14 cells show numbers of IP3R3 clusters in cells treated with control or EB3 siRNA alone or after rescue with EB3-GFP or EB3ΔAc-GFP. Individual data points and mean ± SEM are shown. ∗∗∗Compared to control siRNA-treated cells using one-way ANOVA.
(C) Time-lapse images (collected at 0.5-s intervals) of GFP-IP3R3 expressed in HLMVECs treated with control or EB3 siRNA and stimulated with α-thrombin (50 nM, as indicated). Times (s) are shown in each panel. Scale bar, 10 μm.
(D and E) Summary results (mean ± SEM from 11 cells in each group) show time course of GFP-IP3R3 clustering after addition of α-thrombin (at t = 0) and (E) the lifetime of the clusters (time taken for 90% of clusters to disappear). These analyses were performed using processed images in which clusters present before addition of α-thrombin were subtracted (see Experimental Procedures). ∗∗Using Student’s t test. Depletion of EB3 reduced both the number and lifetime of the IP3R3 clusters. Figure S4 shows additional control experiments and related analyses of GFP-IP3R2.
GFP-IP3R3 also formed clusters in unstimulated HLMVECs, with fewer clusters in cells lacking EB3 (Figure 3C). This allowed dynamic imaging of GFP-IP3R3 distribution in response to receptor activation. Stimulation of HLMVECs expressing GFP-IP3R3 with α-thrombin evoked a rapid transient increase in IP3R3 clustering that peaked after ∼30 s and persisted for 190 ± 15 s (time for 90% of clusters to disappear) (Figures 3C–3E). Similar clustering of IP3Rs in response to stimuli that caused IP3R activation has been reported in other cells (Taufiq-Ur-Rahman et al., 2009, Tateishi et al., 2005). Both the number of IP3R3 clusters after stimulation with α-thrombin and their lifespan (64 ± 28 s) were reduced in EB3-depleted HLMVECs (Figures 3D and 3E).
Residue T804 within the TEIP motif of IP3R3 was required for IP3R3 binding to EB3 (Figure 2C). Expression of GFP-IP3R3(T804A) in HLMVECs attenuated α-thrombin-evoked Ca2+ signals. This was evident from the smaller effects of α-thrombin on both the decrease in [Ca2+]ER and increase in [Ca2+]c in cells expressing GFP-IP3R3(T804A) relative to those expressing GFP-IP3R3 (Figure 4). GFP-IP3R3(T804A) also formed fewer clusters than GFP-IP3R3, they barely responded to α-thrombin, and the few clusters that formed were short lived (Figures 5A–5C). We conclude that EB3, via its interaction with IP3R3 in endothelial cells, both dynamically regulates basal and agonist-evoked clustering of IP3R3 and the IP3-mediated Ca2+ release induced by α-thrombin (Figure 5D).
Figure 4.

Interaction between IP3R3 and EB3 Is Required for α-Thrombin-Induced Ca2+ Signaling
(A) Time-lapse images (times, in s, shown in panels) of HLMVEC expressing GFP-IP3R3 (WT) or GFP-IP3R3(T804A) mutant (green) with the ER luminal Ca2+ indicator, R-CEPIA1er (red) in overlaid images. R-CEPIA1er fluorescence is color coded with warm colors denoting high [Ca2+]ER. Stimulation with α-thrombin (50 nM at t = 0) in Ca2+-free medium caused a decrease in [Ca2+]ER in cells expressing wild-type (WT) GFP-IP3R3, but not in cells expressing GFP-IP3R3(T804A). Scale bar, 10 μm.
(B) Representative traces (normalized to 100% at 20 s before addition of α-thrombin) show [Ca2+]ER monitored with R-CEPIA1er in individual cells expressing GFP-IP3R3 or GFP-IP3R3(T804A) and stimulated with α-thrombin (50 nM) in Ca2+-free medium.
(C) Effects of α-thrombin (50 nM) on [Ca2+]c recorded from ∼5–10 fura-2-loaded HLMVECs expressing GFP-IP3R3 (WT) or GFP-IP3R3(T804A), or adjacent untransfected cells.
(D) Summary results show peak changes in [Ca2+]c (Δ[Ca2+]c) evoked by α-thrombin. Individual data points (four to ten cells) and means ± SEM are plotted. ∗∗∗, ∗∗Using one-way ANOVA.
Figure 5.

Interaction between IP3R3 and EB3 Is Required for Effective IP3R3 Clustering
(A) Time-lapse images (times, in s, shown in panels) of GFP fluorescence in HLMVECs expressing GFP-IP3R3(WT) or GFP-IP3R3(T804A) after stimulation with α-thrombin (50 nM, at t = 0). Scale bar, 10 μm.
(B and C) Summary results (mean ± SEM from five to seven cells) show the number of clusters for GFP-IP3R3 and GFP-IP3R3(T804A) (B) and cluster lifetime (C) after stimulation with α-thrombin (50 nM at t = 0). Data were analyzed using processed images as in Figures 3D and 3E. ∗∗Using Student’s t test.
(D) Model for EB3-dependent IP3R3 clustering and amplification of Ca2+ signals. EB3 facilitates clustering of IP3Rs within ER membranes (left). α-Thrombin, via PAR-1, activates PLC and synthesis of IP3. We propose that IP3Rs more effectively release Ca2+ in response to this IP3 when they have clustered, possibly because amplification of the signals by Ca2+-induced Ca2+ release is more effective.
Loss of EB3 Suppresses Vascular Leakage In Vivo
Ca2+ signals, via both activation of protein kinase C α and myosin light chain (MLC) kinase, facilitate cell contraction and destabilization of AJs (Komarova and Malik, 2010, Vandenbroucke St Amant et al., 2012). We therefore examined the effects of EB3 and IP3R3 on phosphorylation of MLC-II. We used human pulmonary artery endothelial cells (HPAECs) for these analyses because they are amenable to measurements of trans-endothelial electrical resistance (TEER), which directly report, in real time, changes in the integrity of AJs (Vandenbroucke St Amant et al., 2012, Szulcek et al., 2013). Depletion of EB3 or IP3R3 (Figure S4C) suppressed α-thrombin-induced phosphorylation of MLC-II in HPAECs (Figures 6A and 6B). In confluent monolayers of HPAECs, α-thrombin induced a decrease in TEER, reflecting changes in cell shape and disruption of AJs between endothelial cells. The decrease in TEER was attenuated in cells lacking IP3R3 or EB3 (Figures 6C and 6D), consistent with the lesser α-thrombin-evoked phosphorylation of MLC-II in these cells. These findings suggest that the interactions between EB3 and IP3R3, through their effects on α-thrombin-induced Ca2+ signals, contribute to MLC-II activation and the increased permeability of the endothelial barrier.
Figure 6.

Depletion of EB3 Attenuates α-Thrombin-Induced Phosphorylation of MLC-II and Endothelial Hyper-permeability
(A and B) Time course of MLC-II phosphorylation in HPAECs treated with the indicated siRNAs and stimulated with α-thrombin (50 nM at t = 0). The western blots are typical of two to three experiments.
(C) Typical effects of α-thrombin (50 nM) on TEER in HPAECs treated with the indicated siRNAs. Basal resistance was normalized to 1.
(D) Summary results show the maximum change in TEER (ΔTEER) evoked by α-thrombin. Individual data points with mean ± SEM are shown, n = 6–20 per group, ∗, ∗∗∗Compared to control siRNA-treated cells using one-way ANOVA.
(E) Permeability of endothelial vessel wall in lungs from EB3-iECKO and Cre-negative mice assessed by measuring microvascular filtration coefficient, kf,c (see Experimental Procedures). Isolated lungs were infused with 30 μM PAR-1 agonist peptide (PAR1-AP), and kf,c was calculated from the slope of the weight-gain curve between 15 and 20 min after the infusion. Individual data points are shown with mean ± SD, n = 3 mice per group; ∗From ANOVA. See also Figure S5.
Ca2+ signals in the endothelium play a critical role in regulating vascular permeability, a hallmark of acute lung injury and inflammation (Gandhirajan et al., 2013, Tauseef et al., 2012). To determine the role of EB3 in vascular endothelium, we generated EB3-iECKO mice in which high-fidelity inducible deletion of the EB3 gene (Mapre3) was restricted to endothelial cells (Figure S5). We then used lungs to determine the microvessel filtration coefficient (kf,c), a measure of endothelial vascular permeability to liquid, in naive lungs and after activation of PAR-1 (Figure 6E). Lungs from control Tie2-CreERT2-negative and EB3-iECKO mice had similar basal permeability, suggesting that EB3 is not essential in the adult microvasculature (Figure 6E). Infusion of a peptide agonist of PAR-1 (PAR1-AP) caused lung vascular hyper-permeability in wild-type, but not in EB3-iECKO mice (Figure 6E), indicating a pivotal role of EB3 in mediating the lung vascular permeability response.
Discussion
Here, we show that stimulation of PLCβ as induced by the inflammatory mediator α-thrombin causes IP3R3s to assemble into clusters in the ER membrane of endothelial cells and that this event requires the association of the TxIP motif of IP3R3 with EB3 located at the tip of growing microtubules. Disrupting this interaction inhibits Ca2+ signaling, the phosphorylation of MLC-II, and the subsequent increase in endothelial permeability in response to α-thrombin.
The local density of IP3Rs in the ER membrane induced by IP3R clustering is thought to be responsible for the spatially organized nature of Ca2+ signaling in distinct cellular domains (Isshiki et al., 1998, Low et al., 2010). The spacing of IP3Rs is key in determining whether the Ca2+ released stimulates the activity of neighboring IP3Rs, and hence propagates Ca2+ signaling in a regenerative manner. Optimal clustering of IP3Rs is therefore believed to be a critical factor in Ca2+ signaling (Shuai and Jung, 2003). A major factor determining IP3R clustering may be IP3-evoked conformational changes in IP3Rs (Taufiq-Ur-Rahman et al., 2009, Tateishi et al., 2005). Studies also showed that microtubules facilitated movement of IP3Rs in cells (Ferreri-Jacobia et al., 2005), and further that disruption of microtubules inhibited IP3-evoked Ca2+ spiking (Béliveau and Guillemette, 2009, Fogarty et al., 2000), suggesting a role of microtubules in the mechanism of IP3R clustering. However, Taufiq-Ur-Rahman et al. (2009) showed that clustering of IP3Rs within isolated nuclei did not require microtubules. Thus, it has not been resolved whether microtubules regulate ER dynamics, and, if so, by what mechanisms and whether microtubule regulation of IP3R clustering has functional relevance in an important Ca2+-regulated biological response. Our results show the direct interaction of IP3Rs with microtubules via the microtubule tip protein EB3 is required for the assembly of IP3Rs into signaling clusters at the ER membrane, and these clusters mediate Ca2+ release from ER stores in endothelial cells. Our previous work established that IP3-induced Ca2+ release in endothelial cells activated the phosphatase calcineurin, which dephosphorylated EB3, enabling EB3 dimerization. EB3 dimers, in turn, bound microtubules to promote persistent growth and disassembly of AJs (Komarova et al., 2012). The present work demonstrates that the microtubules functioning via EB3 are required for IP3R clustering at the ER membrane and the genesis of intracellular Ca2+ signaling in endothelial cells.
The S/TxIP motif, located immediately downstream of the IP3-binding site and conserved in all mammalian IP3Rs, was required for IP3Rs binding specifically to the C-terminal tail of EB3 (as opposed to EB1) in endothelial cells. The enhanced binding of EB3 to IP3R3 correlated with the requirement for EB3, rather than EB1, for α-thrombin-induced Ca2+ signaling. The requirement for EB3 may also result from its binding to additional, as-yet-unidentified proteins mediating clustering of IP3R3s. Indeed, residues surrounding the S/TxIP motif have been recently shown to recognize EB3 (Leśniewska et al., 2014). Inhibition of α-thrombin-induced Ca2+ signals by a fragment of EB3 (Ct-mRFP) that dimerizes with native EB3 and prevents binding to microtubules confirmed that the requirement for EB3 is associated with its ability to bind to the growing microtubule tips. The concept of EB3 binding to IP3R3 is further supported by our FRET analysis showing a close and specific apposition of IP3R3 and EB3 at microtubule tips.
We found that IP3R3 and IP3R2 are the major IP3R subtypes in endothelial cells. IP3R2s are concentrated in perinuclear regions and clustered in unstimulated endothelial cells. The perinuclear distribution of IP3R2 (Pantazaka and Taylor, 2011) and their propensity to cluster in unstimulated cells are also observed in other cells (Iwai et al., 2005, Sheppard et al., 1997). The latter finding suggests that basal levels of IP3 are sufficient to stimulate IP3R2 clustering, consistent with the greater affinity of IP3R2 for IP3 relative to other subtypes (Iwai et al., 2007). In unstimulated endothelial cells, however, IP3R3s are more widely distributed and less clustered than IP3R2s. The key role of IP3R3 in mediating α-thrombin-induced Ca2+ signals is therefore probably due to their proximity to IP3 produced by PLC at the plasma membrane. Our observation that α-thrombin augments the clustering of IP3R3s is consistent with their lower sensitivity to IP3 (Iwai et al., 2007). IP3-induced clustering of IP3R3s in the nuclear envelope did not require microtubules (Taufiq-Ur-Rahman et al., 2009), and yet in endothelial cells clustering requires EB3-mediated interaction of IP3R3 with microtubules. The discrepancy may reflect a need for microtubules to facilitate movement of IP3Rs within the crowded cytoplasm of intact cells and stabilize IP3R clusters once they have formed.
We conclude that EB3-mediated tethering of IP3R3s to microtubule tips in endothelial cells is required to assemble the Ca2+ signaling machinery. Moreover, α-thrombin stimulates formation of IP3 at the plasma membrane, where it facilitates the clustering of IP3R3 via EB3-mediated interaction with microtubules. We propose that IP3R3 clustering enables Ca2+-mediated amplification of IP3-induced Ca2+ release through activation of neighboring IP3Rs. The IP3-evoked Ca2+ signaling can therefore be attenuated in the absence of this amplification step. The physiological importance of the EB3-IP3R3 interaction is evident from results in lung microvessels showing that endothelial cell-specific knockout of EB3 prevents the increase in endothelial permeability in response to inflammatory signal. Thus, microtubule-associated EB3 interacts directly with IP3Rs to assemble the Ca2+ signaling complex at the ER membrane, and disruption of EB3-IP3R interaction may be an attractive therapeutic target for vascular inflammation.
Experimental Procedures
Materials
Sources of the expression constructs, antibodies, and siRNAs are provided in Supplemental Experimental Procedures. mRFP-(rat)CLIP-170 was generated from EGFP-CLIP-170 (Komarova et al., 2005) by substituting EGFP with monomeric RFP. For EB3-Ct-mRFP, the C terminus (residues 200–281) of EB3 was amplified by PCR and sub-cloned into the pmRFP-N1 vector (a gift from Dr. R. Tsien) at Sal1 and BamH1 sites. Expression constructs for the siRNA-insensitive form of EB3 and for EGFP-IP3R3(T804A) were generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies).
DAPI was from Sigma. Human α-thrombin was from Fisher Scientific. The PAR-1-activating peptide (PAR1-AP, TFLLRN-NH2, ∼90% purity) was synthesized by the Research Resources Core at UIC. Sources of other materials are provided in the relevant sections in Experimental Procedures.
Pull-Down Assays with EB Proteins
Preparation of bacterially expressed (His6)-tagged EB proteins and their covalent immobilization on Ni-NTA columns for pull-down experiments using lysates from HEK cells expressing GFP-tagged proteins are described in Supplemental Experimental Procedures.
Cell Culture and Transfections
HPAECs and HLMVECs (Lonza) were grown in EGM-2 medium supplemented with 10% fetal bovine serum (FBS) and EGM-2 MV Bulletkit or EGM-2 Bulletkit (Lonza), respectively. Cells were used between passages 2 and 6. CHO-K1 and HEK293 cells (ATCC) were grown in DMEM with 10% FBS (Gibco). Cells were transfected at ∼80% confluence using X-tremeGENE HP according to the manufacturer’s protocol (Roche) and used after 24–48 hr. For siRNA-mediated inhibition of protein expression, cells were treated with 70 nM siRNA (Supplemental Experimental Procedures) using GeneSilencer transfection reagent according to the manufacturer’s protocol (Genlantis) and used after 72–96 hr. For experiments in which cells were stimulated with α-thrombin, the FBS concentration of the culture medium was reduced to 0.5% for 1 hr before the experiment.
Measurements of [Ca2+]c
HLMVECs grown on glass-bottomed dishes (Becton Dickinson) were loaded with fura-2 AM (3 μM, Life Technologies) for 20 min at 37°C in culture medium without supplements. The medium was then replaced with medium comprising: 150 mM NaCl, 4 mM KCl, 1 mM MgCl2, 5.6 mM glucose, and 25 mM HEPES (pH 7.4), and, after ∼10 min, cells were used for experiments at 25°C. Fura-2 fluorescence was excited at 340 and 380 nm and collected at 510 ± 80 nm using an Axiovert 100 inverted microscope (Carl Zeiss) equipped with Plan-Apo 60× with the numerical aperture (NA) 1.4 oil immersion objective, Lambda DG-4 switcher illumination system (Sutter Instruments), AxioCom Hsm camera (Zeiss), fura-2 filter set (Chroma), and AxioVision Physiology Acquisition module. Images were collected at 2-s intervals. Fluorescence ratios (F340/F380) were calculated within a circular region of interest (radius 3 μm) for each cell after subtraction of intracellular background fluorescence, determined by quenching fura-2 fluorescence by addition of 3 μM ionomycin with 5 mM MnCl2. [Ca2+]c was calculated from F340/F380 ratios by reference to Ca2+ standard solutions (Life Technologies). Measurements of cytosolic IP3 concentrations are described in Supplemental Experimental Procedures.
Measurements of [Ca2+]ER
The free [Ca2+] within the ER ([Ca2+]ER) was measured in HLMVECs transfected with the GEM-CEPIA1er ratiometric indicator (Suzuki et al., 2014). Analyses were performed at 37°C in the Ca2+-free medium described above using a confocal microscope (Zeiss LSM 710, Axio Observer Z1) equipped with a 63× 1.4 NA Plan-Apochromat oil immersion objective and a diode-pumped solid-state laser. Cells were excited at 405 nm, and emission was collected by two photomultiplier tubes (456–476 and 510–530 nm). Images (5–10/cell) were collected at 1-s intervals, averaged over four frames to optimize signal-to-noise ratios, and fluorescence ratios (R = F466/F510) were calculated. For calibration, the plasma membrane was permeabilized (150 μM β-escin, 4 min), Rmin and Rmax were then determined in the absence of β-escin in medium with (Rmax) or without (Rmin) Ca2+. The medium comprised: 140 mM KCl, 10 mM NaCl, 1 mM MgCl2, 20 mM HEPES, 3 μM ionomycin, and 3 μM thapsigargin (pH 7.4), and either 0.3 mM EGTA (Rmin) or 10 mM CaCl2 (Rmax). [Ca2+]ER was then calculated from the observed fluorescence ratio (R):
where n, Hill coefficient (1.37) and KD for Ca2+ = 558 μM (Suzuki et al., 2014).
A confocal microscope (Zeiss, LSM 710, Axio Observer Z1) with BIG detector was used to capture dual-color images of m-Cherry-er with G-CEPIAer. For comparisons of relative changes in [Ca2+]ER (Figure 1D), a cross-sectional view of the intensity values of G-CEPIAer fluorescence over time was generated, and the relative changes in [Ca2+]ER were calculated.
Immunofluorescence and Live-Cell Imaging
HLMVECs were fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100, and immunostained for IP3R2 or IP3R3 (Supplemental Experimental Procedures). All z stack confocal images (Zeiss LSM 510 META) were acquired with the same settings to allow comparisons of IP3R distributions between treatment groups. A projected image was generated by collecting a maximum voxel value through each z stack.
The methods used to quantify microtubule dynamics using EB1-GFP, assess the interactions between GFP-IP3R3 and EB3-mRFP or mRFP-CLIP-170 using acceptor photobleaching, and to quantify clustering of GFP-IP3R3 or immunostained IP3R2 and IP3R3 are described in Supplemental Experimental Procedures.
Trans-endothelial Electric Resistance Measurements
HPAECs were plated onto gelatin-coated 8W1E gold electrodes (Applied Biophysics), transfected with siRNA, and used after 72 hr (Garcia et al., 2011). Changes in trans-endothelial electric resistance (TEER) in response to α-thrombin were monitored using an Electric Cell Substrate Impedance Sensing system (Applied Biophysics) and normalized to the basal resistance.
Measurement of Vessel Filtration Coefficient (kfc)
Animal care and handling were performed according to an approved protocol of the University of Illinois at Chicago Animal Care Committee. The transgenic mice are described in Supplemental Experimental Procedures. For measurements of kf,c, isolated lungs were perfused with RPMI medium at constant flow (2 ml/min), temperature (37°C), and venous pressure (4 cm H2O) as previously described (Garcia et al., 2011). The preparation was ventilated at 120 breaths/min with constant peak inspiratory (∼10 cm H2O) and end expiratory pressures (2 cmH2O). Lung weight change was recorded with a force-displacement transducer (Model FT03C, Grass Technologies). A 20-min equilibration perfusion established isogravimetric conditions, before measuring the gravimetric filtration coefficient (kf,c) by comparing the rate of lung weight-gain during a baseline isogravimetric period with the rate after a change in hydrostatic pressure of at least 6 cm H2O for 20 min. The filtration rate was determined from the slope of the weight-gain curve between 15 and 20 min after PAR1-AP infusion. kf,c was calculated from:
where W, weight; t, time; P, pulmonary microvascular pressure.
Endothelial lysates were collected after each measurement of kf,c via a left atrial cannula perfused with buffer (50 mM Tris-Cl (pH 7.8), 0.2% Triton X-100, and protease and phosphatase inhibitor cocktails; 0.4 ml/min). Fractions were collected at 1-min intervals. Fractions 2 and 3, which were positive for VE-cadherin (endothelial marker) and negative for smooth muscle actin, were used for assessment of EB3 expression by western blot.
Statistical Analyses
Comparisons between groups were made using ANOVA with the Tukey post-test method or Student’s t test. Significance values are shown by ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Author Contributions
Y.A.K. conceived the project; M.G., F.H., Y.S., S.M.V., and Y.A.K. conducted experiments; M.G., C.W.T., and Y.A.K. analyzed and interpreted data; M.G., C.W.T., A.B.M., and Y.A.K. wrote the paper.
Acknowledgments
We thank the Core Imaging Facility of the Research Resources Center, University of Illinois at Chicago for use of microscopes. We thank Dr. A. Tanimura (Hokkaido, Japan) for LIBRAvIII and GFP-IP3R3; Dr. M. Iino (Tokyo, Japan) for G-CEPIA1er, R-CEPIA1er, GEM-CEPIA1er, and mCherry-er; Dr. R. Tsien (UCSD, CA) for the pmRFP-N1 vector; Drs. P. Chambon (IGBMC, France) and S. Offermanns (Max Planck Institute, Germany) for the tie2-CreERT2 mouse line; Dr. S. Muallem (NIDCR, Bethesda) for YFP-STIM1; Dr. A. Akhmanova (Utrecht University, the Netherlands) for mRFP-CLIP-170. Supported by NIH grants R01 HL103922 and Giles F. Filley Memorial Award to Y.A.K.; PO1 HL60678 to A.B.M.; T32 HL07829-17 and AHA AWARD 13PRE17090090 to M.G., and a Wellcome Trust Senior Investigator Award (101844) to C.W.T. The work presented fulfills in part the PhD degree requirement for M.G. Y.A.K., S.M.V., and A.B.M. have a patent that is potentially related to this study.
Published: June 25, 2015
Footnotes
This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, five figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.06.001.
Supplemental Information
References
- Akhmanova A., Steinmetz M.O. Microtubule +TIPs at a glance. J. Cell Sci. 2010;123:3415–3419. doi: 10.1242/jcs.062414. [DOI] [PubMed] [Google Scholar]
- Béliveau E., Guillemette G. Microfilament and microtubule assembly is required for the propagation of inositol trisphosphate receptor-induced Ca2+ waves in bovine aortic endothelial cells. J. Cell. Biochem. 2009;106:344–352. doi: 10.1002/jcb.22011. [DOI] [PubMed] [Google Scholar]
- Chalmers M., Schell M.J., Thorn P. Agonist-evoked inositol trisphosphate receptor (IP3R) clustering is not dependent on changes in the structure of the endoplasmic reticulum. Biochem. J. 2006;394:57–66. doi: 10.1042/BJ20051130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshjou N., Sieracki N., van Nieuw Amerongen G.P., Conway D.E., Schwartz M.A., Komarova Y.A., Malik A.B. Rac1 functions as a reversible tension modulator to stabilize VE-cadherin trans-interaction. J. Cell Biol. 2015;208:23–32. doi: 10.1083/jcb.201409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E. Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- Ferreri-Jacobia M., Mak D.O., Foskett J.K. Translational mobility of the type 3 inositol 1,4,5-trisphosphate receptor Ca2+ release channel in endoplasmic reticulum membrane. J. Biol. Chem. 2005;280:3824–3831. doi: 10.1074/jbc.M409462200. [DOI] [PubMed] [Google Scholar]
- Fogarty K.E., Kidd J.F., Turner A., Skepper J.N., Carmichael J., Thorn P. Microtubules regulate local Ca2+ spiking in secretory epithelial cells. J. Biol. Chem. 2000;275:22487–22494. doi: 10.1074/jbc.M909402199. [DOI] [PubMed] [Google Scholar]
- Foskett J.K., White C., Cheung K.H., Mak D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J.R., Voeltz G.K. The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol. 2011;21:709–717. doi: 10.1016/j.tcb.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhirajan R.K., Meng S., Chandramoorthy H.C., Mallilankaraman K., Mancarella S., Gao H., Razmpour R., Yang X.F., Houser S.R., Chen J. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Invest. 2013;123:887–902. doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A.N., Vogel S.M., Komarova Y.A., Malik A.B. Permeability of endothelial barrier: cell culture and in vivo models. Methods Mol. Biol. 2011;763:333–354. doi: 10.1007/978-1-61779-191-8_23. [DOI] [PubMed] [Google Scholar]
- Giannotta M., Trani M., Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev. Cell. 2013;26:441–454. doi: 10.1016/j.devcel.2013.08.020. [DOI] [PubMed] [Google Scholar]
- Graier W.F., Paltauf-Doburzynska J., Hill B.J., Fleischhacker E., Hoebel B.G., Kostner G.M., Sturek M. Submaximal stimulation of porcine endothelial cells causes focal Ca2+ elevation beneath the cell membrane. J. Physiol. 1998;506:109–125. doi: 10.1111/j.1469-7793.1998.109bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I., Gouveia S.M., van der Vaart B., Demmers J., Smyth J.T., Honnappa S., Splinter D., Steinmetz M.O., Putney J.W., Jr., Hoogenraad C.C., Akhmanova A. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 2008;18:177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honnappa S., Gouveia S.M., Weisbrich A., Damberger F.F., Bhavesh N.S., Jawhari H., Grigoriev I., van Rijssel F.J., Buey R.M., Lawera A. An EB1-binding motif acts as a microtubule tip localization signal. Cell. 2009;138:366–376. doi: 10.1016/j.cell.2009.04.065. [DOI] [PubMed] [Google Scholar]
- Howard J., Hyman A.A. Dynamics and mechanics of the microtubule plus end. Nature. 2003;422:753–758. doi: 10.1038/nature01600. [DOI] [PubMed] [Google Scholar]
- Isshiki M., Ando J., Korenaga R., Kogo H., Fujimoto T., Fujita T., Kamiya A. Endothelial Ca2+ waves preferentially originate at specific loci in caveolin-rich cell edges. Proc. Natl. Acad. Sci. USA. 1998;95:5009–5014. doi: 10.1073/pnas.95.9.5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai M., Tateishi Y., Hattori M., Mizutani A., Nakamura T., Futatsugi A., Inoue T., Furuichi T., Michikawa T., Mikoshiba K. Molecular cloning of mouse type 2 and type 3 inositol 1,4,5-trisphosphate receptors and identification of a novel type 2 receptor splice variant. J. Biol. Chem. 2005;280:10305–10317. doi: 10.1074/jbc.M413824200. [DOI] [PubMed] [Google Scholar]
- Iwai M., Michikawa T., Bosanac I., Ikura M., Mikoshiba K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2007;282:12755–12764. doi: 10.1074/jbc.M609833200. [DOI] [PubMed] [Google Scholar]
- Komarova Y., Malik A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- Komarova Y., Lansbergen G., Galjart N., Grosveld F., Borisy G.G., Akhmanova A. EB1 and EB3 control CLIP dissociation from the ends of growing microtubules. Mol. Biol. Cell. 2005;16:5334–5345. doi: 10.1091/mbc.E05-07-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova Y., De Groot C.O., Grigoriev I., Gouveia S.M., Munteanu E.L., Schober J.M., Honnappa S., Buey R.M., Hoogenraad C.C., Dogterom M. Mammalian end binding proteins control persistent microtubule growth. J. Cell Biol. 2009;184:691–706. doi: 10.1083/jcb.200807179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova Y.A., Huang F., Geyer M., Daneshjou N., Garcia A., Idalino L., Kreutz B., Mehta D., Malik A.B. VE-cadherin signaling induces EB3 phosphorylation to suppress microtubule growth and assemble adherens junctions. Mol. Cell. 2012;48:914–925. doi: 10.1016/j.molcel.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leśniewska K., Warbrick E., Ohkura H. Peptide aptamers define distinct EB1- and EB3-binding motifs and interfere with microtubule dynamics. Mol. Biol. Cell. 2014;25:1025–1036. doi: 10.1091/mbc.E13-08-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J.T., Shukla A., Behrendorff N., Thorn P. Exocytosis, dependent on Ca2+ release from Ca2+ stores, is regulated by Ca2+ microdomains. J. Cell Sci. 2010;123:3201–3208. doi: 10.1242/jcs.071225. [DOI] [PubMed] [Google Scholar]
- Maurer S.P., Fourniol F.J., Bohner G., Moores C.A., Surrey T. EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell. 2012;149:371–382. doi: 10.1016/j.cell.2012.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuyama F., Sawai T. The redistribution of Ca2+ stores with inositol 1,4,5-trisphosphate receptor to the cleavage furrow in a microtubule-dependent manner. Int. J. Dev. Biol. 2001;45:861–868. [PubMed] [Google Scholar]
- Pantazaka E., Taylor C.W. Differential distribution, clustering, and lateral diffusion of subtypes of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 2011;286:23378–23387. doi: 10.1074/jbc.M111.236372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendin D., McNew J.A., Daga A. Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr. Opin. Cell Biol. 2011;23:435–442. doi: 10.1016/j.ceb.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo-Guisado E., Campbell D.G., Deak M., Alvarez-Barrientos A., Morrice N.A., Alvarez I.S., Alessi D.R., Martín-Romero F.J. Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J. Cell Sci. 2010;123:3084–3093. doi: 10.1242/jcs.067215. [DOI] [PubMed] [Google Scholar]
- Ribeiro C.M., Reece J., Putney J.W., Jr. Role of the cytoskeleton in calcium signaling in NIH 3T3 cells. An intact cytoskeleton is required for agonist-induced [Ca2+]i signaling, but not for capacitative calcium entry. J. Biol. Chem. 1997;272:26555–26561. doi: 10.1074/jbc.272.42.26555. [DOI] [PubMed] [Google Scholar]
- Sheppard C.A., Simpson P.B., Sharp A.H., Nucifora F.C., Ross C.A., Lange G.D., Russell J.T. Comparison of type 2 inositol 1,4,5-trisphosphate receptor distribution and subcellular Ca2+ release sites that support Ca2+ waves in cultured astrocytes. J. Neurochem. 1997;68:2317–2327. doi: 10.1046/j.1471-4159.1997.68062317.x. [DOI] [PubMed] [Google Scholar]
- Shuai J.W., Jung P. Optimal ion channel clustering for intracellular calcium signaling. Proc. Natl. Acad. Sci. USA. 2003;100:506–510. doi: 10.1073/pnas.0236032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I.F., Wiltgen S.M., Parker I. Localization of puff sites adjacent to the plasma membrane: functional and spatial characterization of Ca2+ signaling in SH-SY5Y cells utilizing membrane-permeant caged IP3. Cell Calcium. 2009;45:65–76. doi: 10.1016/j.ceca.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J., Kanemaru K., Ishii K., Ohkura M., Okubo Y., Iino M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014;5:4153. doi: 10.1038/ncomms5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulcek R., Beckers C.M., Hodzic J., de Wit J., Chen Z., Grob T., Musters R.J., Minshall R.D., van Hinsbergh V.W., van Nieuw Amerongen G.P. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc. Res. 2013;99:471–482. doi: 10.1093/cvr/cvt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K., Shin R.M., Inoue T., Kato K., Mikoshiba K. Regulation of nerve growth mediated by inositol 1,4,5-trisphosphate receptors in growth cones. Science. 1998;282:1705–1708. doi: 10.1126/science.282.5394.1705. [DOI] [PubMed] [Google Scholar]
- Tanimura A., Morita T., Nezu A., Shitara A., Hashimoto N., Tojyo Y. Use of fluorescence resonance energy transfer-based biosensors for the quantitative Aanalysis of inositol 1,4,5-trisphosphate dynamics in calcium oscillations. J. Biol. Chem. 2009;284:8910–8917. doi: 10.1074/jbc.M805865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaka K., Mio M., Fujisawa K., Aoki I. Role of microtubules on Ca2+ release from the endoplasmic reticulum and associated histamine release from rat peritoneal mast cells. Biochem. Pharmacol. 1991;41:1031–1037. doi: 10.1016/0006-2952(91)90211-m. [DOI] [PubMed] [Google Scholar]
- Tateishi Y., Hattori M., Nakayama T., Iwai M., Bannai H., Nakamura T., Michikawa T., Inoue T., Mikoshiba K. Cluster formation of inositol 1,4,5-trisphosphate receptor requires its transition to open state. J. Biol. Chem. 2005;280:6816–6822. doi: 10.1074/jbc.M405469200. [DOI] [PubMed] [Google Scholar]
- Taufiq-Ur-Rahman, Skupin A., Falcke M., Taylor C.W. Clustering of InsP3 receptors by InsP3 retunes their regulation by InsP3 and Ca2+ Nature. 2009;458:655–659. doi: 10.1038/nature07763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauseef M., Knezevic N., Chava K.R., Smith M., Sukriti S., Gianaris N., Obukhov A.G., Vogel S.M., Schraufnagel D.E., Dietrich A. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012;209:1953–1968. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C.W., Tovey S.C., Rossi A.M., Lopez Sanjurjo C.I., Prole D.L., Rahman T. Structural organization of signalling to and from IP3 receptors. Biochem. Soc. Trans. 2014;42:63–70. doi: 10.1042/BST20130205. [DOI] [PubMed] [Google Scholar]
- Tran C.H., Kurjiaka D.T., Welsh D.G. Emerging trend in second messenger communication and myoendothelial feedback. Front Physiol. 2014;5:243. doi: 10.3389/fphys.2014.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke St Amant E., Tauseef M., Vogel S.M., Gao X.P., Mehta D., Komarova Y.A., Malik A.B. PKCα activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ. Res. 2012;111:739–749. doi: 10.1161/CIRCRESAHA.112.269654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermassen E., Van Acker K., Annaert W.G., Himpens B., Callewaert G., Missiaen L., De Smedt H., Parys J.B. Microtubule-dependent redistribution of the type-1 inositol 1,4,5-trisphosphate receptor in A7r5 smooth muscle cells. J. Cell Sci. 2003;116:1269–1277. doi: 10.1242/jcs.00354. [DOI] [PubMed] [Google Scholar]
- Vogel S.M., Malik A.B. Cytoskeletal dynamics and lung fluid balance. Compr Physiol. 2012;2:449–478. doi: 10.1002/cphy.c100006. [DOI] [PubMed] [Google Scholar]
- Wainwright M.S., Rossi J., Schavocky J., Crawford S., Steinhorn D., Velentza A.V., Zasadzki M., Shirinsky V., Jia Y., Haiech J. Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc. Natl. Acad. Sci. USA. 2003;100:6233–6238. doi: 10.1073/pnas.1031595100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B.S., Pfeiffer J.R., Smith A.J., Oliver J.M., Oberdorf J.A., Wojcikiewicz R.J. Calcium-dependent clustering of inositol 1,4,5-trisphosphate receptors. Mol. Biol. Cell. 1998;9:1465–1478. doi: 10.1091/mbc.9.6.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.M., Covington E.D., Lewis R.S. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum-plasma membrane junctions. Mol. Biol. Cell. 2014;25:3672–3685. doi: 10.1091/mbc.E14-06-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.F., Forscher P. Rac1 modulates stimulus-evoked Ca(2+) release in neuronal growth cones via parallel effects on microtubule/endoplasmic reticulum dynamics and reactive oxygen species production. Mol. Biol. Cell. 2009;20:3700–3712. doi: 10.1091/mbc.E08-07-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.