

Abstract
Chondrocytes in the epiphyseal cartilage undergo terminal differentiation prior to their removal through apoptosis. To examine the role of ERK1 and ERK2 in chondrocyte terminal differentiation, we generated Osx-Cre; ERK1−/−; ERK2flox/flox mice (cKOosx), in which ERK1 and ERK2 were deleted in hypertrophic chondrocytes. These cKOosx mice were grossly normal in size at birth, but by 3 weeks of age exhibited shorter long bones. Histological analysis in these mice revealed that the zone of hypertrophic chondrocytes in the growth plate was markedly expanded. In situ hybridization and quantitative real-time PCR analyses demonstrated that Mmp13 and Osteopontin expression was significantly decreased, indicating impaired chondrocyte terminal differentiation. Moreover, Egr1 and Egr2, transcription factors whose expression is restricted to the last layers of hypertrophic chondrocytes in wild type mice, were also strongly downregulated in these cKOosx mice. In transient transfection experiments in the RCS rat chondrosarcoma cell line, the expression of Egr1, Egr2, or a constitutively active mutant of MEK1 increased the activity of an Osteopontin promoter, while the MEK1-induced activation of the Osteopontin promoter was inhibited by the co-expression of Nab2, an Egr1 and Egr2 co-repressor. These results suggest that MEK1-ERK signaling activates the Osteopontin promoter in part through Egr1 and Egr2. Finally, our histological analysis of cKOosx mice demonstrated enchondroma-like lesions in the bone marrow that are reminiscent of human metachondromatosis, a skeletal disorder caused by mutations in PTPN11. Our observations suggest that the development of enchondromas in metachondromatosis may be caused by reduced ERK MAPK signaling.
Keywords: ERK, Hypertrophic chondrocytes, Terminal differentiation, Endochondral ossification, Metachondromatosis
Introduction
In vertebrates, long bones are formed through endochondral ossification, in which chondrocytes undergo a series of proliferation and differentiation processes (1). This process begins when undifferentiated mesenchymal cells condense and differentiate into chondrocytes. Cells in the chondrocyte lineage then give rise to distinct zones in the epiphyseal cartilage, beginning with the zones of resting chondrocytes, then proliferating chondrocytes, followed by hypertrophic chondrocytes. Chondrocytes in the zones of resting and proliferating chondrocytes express collagen type II (Col2a1), while chondrocytes in the zone of hypertrophic chondrocytes express collagen type X (Col10a1). Further maturation of hypertrophic chondrocytes into terminally differentiated chondrocytes is characterized by the downregulation of Col10a1 and upregulation of terminal differentiation markers Osteopontin and Matrix metalloproteinase-13 (Mmp13), which are also markers of osteoblasts(1). Ossification occurs when terminally differentiated chondrocytes undergo apoptosis and the calcified cartilage is invaded by blood vessels along with osteoclasts, osteoblasts, and mesenchymal precursor cells.
The entire process of chondrocyte differentiation is under the concerted regulation by multiple signaling pathways(1). One of the key signaling pathways involved in these processes is extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK MAPK), which can be activated by various stimuli, including a number of growth factors and cytokines(2). Various clinical investigations have implicated the critical roles of the ERK MAPK pathway in human skeletal disorders(3). Our previous studies using genetically engineered mice have also indicated that ERK MAPK signaling plays multiple roles at successive steps of chondrocyte differentiation. Overexpression of a constitutively active mutant of MEK1 in undifferentiated mesenchymal cells inhibited the formation of cartilage anlagen(4), indicating that ERK MAPK signaling is inhibitory to early chondrocyte differentiation. In addition, the expression of a constitutively active mutant of MEK1 in chondrocytes under the regulatory sequences of Col2a1 inhibited hypertrophic chondrocyte differentiation(5). The inhibitory effects of ERK MAPK signaling on early chondrocyte differentiation and hypertrophic chondrocyte differentiation have been also demonstrated by ablating ERK1/2 using the Prx1-Cre and Col2a1-Cre transgenes(4). However, the role of ERK MAPK signaling in the terminal differentiation of hypertrophic chondrocytes requires further investigation. In the current study, we examined the roles of ERK1 and ERK2 in terminal chondrocyte differentiation by inactivating ERK1/2 in hypertrophic chondrocytes using the Osx-Cre transgene, which has been shown to direct Cre recombinase activity in hypertrophic chondrocytes in the growth plate in addition to osteoblasts(6-8).
Materials and Methods
Mouse line and breeding
All animal protocols have been approved by the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University. ERK1-null mice(4), mice with the floxed ERK2 allele(4), Osx-Cre transgenic mice(6), and Col1a1CreER-DsRed transgenic mice(9) were described previously. For inducing Cre recombinase activity in Cola1CreER-DsRed embryos, one milligram of tamoxifen (Sigma) dissolved in ethanol and corn oil was injected into the intraperitoneal cavity of the pregnant mother at embryonic day 13.5 (E13.5), E14.5 and E15.5, and embryos were harvested for histological analysis at E18.5.
Tissue preparation and histological analysis
Skeletal preparations were stained with alcian blue and alizarin red as described(4). For histology, tissues were fixed in 10% formalin overnight and embedded in paraffin. Postnatal tissues were demineralized in 0.5M EDTA before embedding. Sections were cut in 7 μm and stained with hematoxylin, eosin, and alcian blue, Masson's trichrome stain, or von Kossa's stain using standard protocols. X-gal staining was performed as described previously(10). Tartrate-resistant acid phosphatase (TRAP) activity was detected using Acid Phosphatase Leukocyte kit (Sigma). TRAP-positive cells were counted along the chondro-osseous junction defined as an area between lines 100 μm above and 100 μm below the junction. Immunostaining was performed using primary antibodies for von Willebrand factor (vWF) (AB7356, dilution 1:1600, Millipore, Billerica, MA), Mmp9 (AB19047, dilution 1:1000, Billerica, MA), ERK1/2 (K23, dilution 1:50, Santa Cruz Biotechnology, Dallas, TX), Egr1 (#4153, dilution 1:100,Cell Signaling, Danvers, MA), Egr2 (PRB236P, dilution 1:200, Covance, New Jersey, NY) and SuperPicture Polymer Detection Kit (#879263, Invitrogen, Carlsbad, CA). Color was developed using ImmPACT DAB (Vector, Burlingame, CA) or TrueBlue (KPL, Gaithersburg, MD). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed with ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Millipore). For bromodeoxyuridine (BrdU) labeling, pregnant mice were intraperitoneally injected with BrdU labeling reagent (3 mg/ml, Invitrogen) at 30 μg/g body weight. Embryos were harvested 4 h after injection, and BrdU incorporation was detected using the BrdU staining kit (Invitrogen). Cell proliferation was quantified by calculating the ratio of BrdU-labeled chondrocytes relative to total number of chondrocytes in the zones of resting and proliferative chondrocytes. RNA in situ hybridization was done using a digoxigenin-labeled riboprobe (Supplemental Table 1). Signal was detected using anti-digoxigenin-alkaline phosphatase (Roche) and NBT/BCIP. All images were taken with a Leica DC500 digital camera with either Leica DM 6000B, DM IRB, or MZ16 microscope using Leica Application Suite 1.3 software.
Quantitative Real-time PCR
The whole tibiae were carefully dissected from E15.5 embryos under a Zeiss Stemi DV4 Stereo microscope. All surrounding connective tissues including perichondrium were removed. Total RNA was extracted using RNeasy kit (Qiagen) and subjected to real-time PCR analysis using TaqMan assays (Applied Biosystems) as described previously(4). Each reaction was performed in triplicate and repeated on at least three independent samples per genotype.
Plasmids, Cell Culture, and Transient Transfection
OPN(−1206)-luc(11) and pcDNA3-Egr1(12) were obtained from Addgene. −914/+5 OPN-luc construct was kindly provided by Martha J. Somerman(13). Egr2 and Nab2 expression plasmids were kind gifts from John Svaren(14). The cDNA for a constitutively active mutant of MEK1 (S218/222E, Δ32-51)(15) was cloned in pcDNA3.1/Zeo (Invitrogen). Transient transfection experiments were performed in rat chondrosarcoma (RCS) cells(16) using GenJet in vitro DNA transfection reagent (SignaGen). Cells were transfected with pRL-SV40 (Promega) and firefly luciferase reporter construct at a 1:99 ratio. Firefly and renilla luciferase activities were assayed with the dual luciferase assay system (Promega) at 48 h after transfection. Firefly luciferase activity was normalized by renilla luciferase activity.
Statistical analysis
Data were calculated from 3-5 independent experiments, and presented as mean ± standard error. Statistical analysis was performed using the student's t-test or one-way analysis of variance. p≤0.05 is considered to be statistically significant.
Results
ERK1/2 inactivation by Osx-Cre delays bone growth
To determine the potential effects of the Osx-Cre transgene per se on bone growth as suggested by other studies(17), we first compared bone length of Osx-Cre; ERK1+/+; ERK2+/+and Osx-Cre; ERK1−/−; ERK2+/flox mice with that of ERK1−/−; ERK2flox/flox mice that do not show any skeletal phenotype(4). The analysis showed that the long bones of Osx-Cre; ERK1+/+; ERK2+/+ and Osx-Cre; ERK1−/−; ERK2+/flox (Control) mice were slightly shorter than those of ERK1−/−; ERK2flox/flox (WT-like) mice at 3 weeks of age, but there were no significant differences between Osx-Cre; ERK1+/+; ERK2+/+ mice and Osx-Cre; ERK1−/−; ERK2+/flox mice (Fig. 1B, data not shown), indicating that the Osx-Cre transgene by itself negatively affects bone growth. Thus, we used Osx-Cre; ERK1−/−; ERK2+/flox mice as a control for the subsequent study.
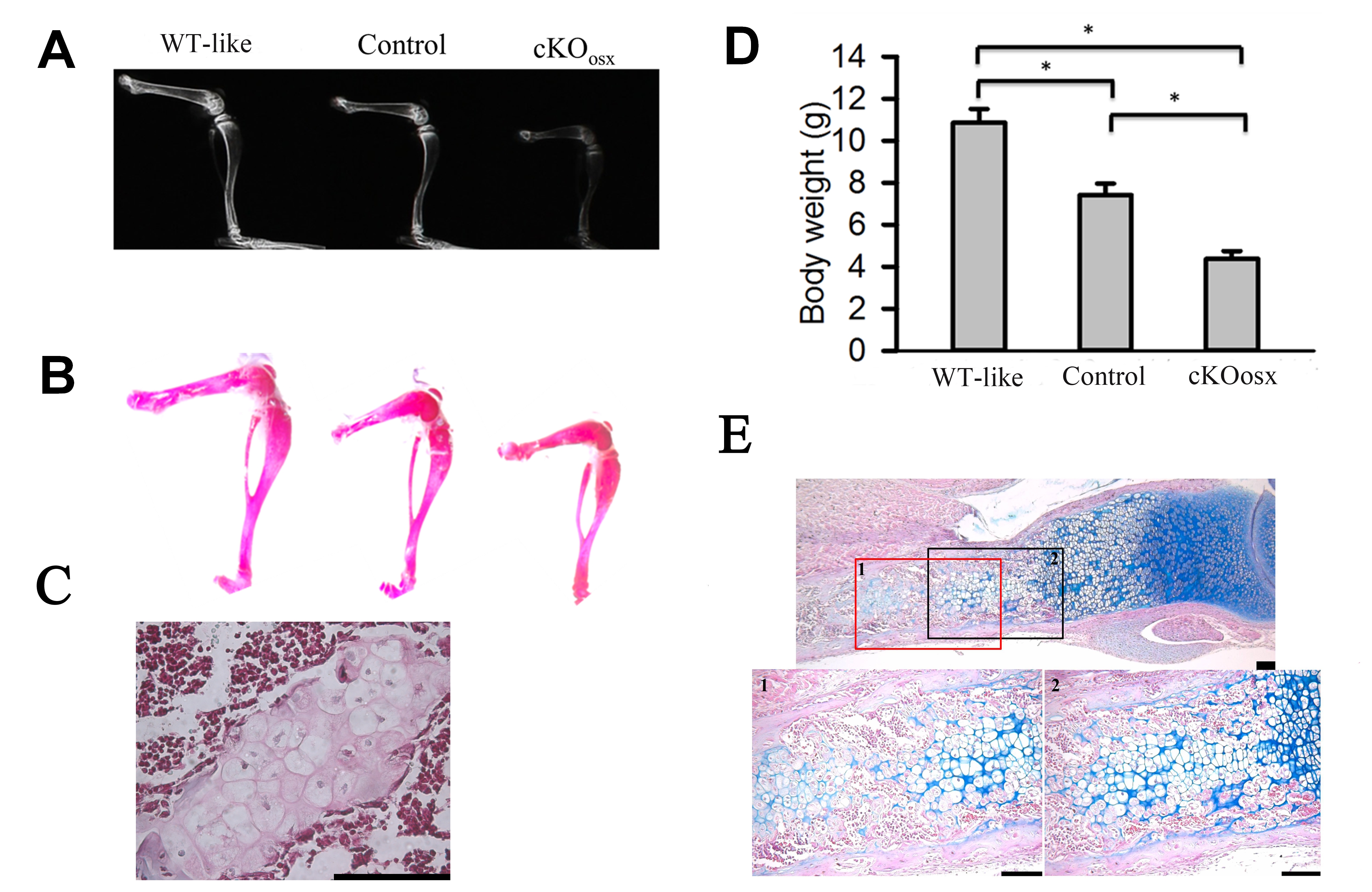
Figure 1. ERK1/2 inactivation using the Osx-Cre transgene causes growth plate defects and delays bone growth.

(A) Osx-Cre; ERK1−/−, ERK2flox/flox (cKOosx) mice showed smaller body size compared with ERK1−/−; ERK2flox/flox (WT-like) and Osx-Cre; ERK1−/−; ERK2+/flox (Control) mice at 3 weeks. (B) Femur and tibia of cKOosx mice were significantly shorter than those of WT-like and Control mice at 3 weeks. Data represent mean ± standard error, n=5 per group. *, p<0.05. NS, not significant. (C) Alcian blue and HE staining of cKOosx and control tibiae showed an expansion of hypertrophic zone in cKOosx mice at E16.5, E17.5, E18.5 and P0. CON, control mice; cKO, cKOosx mice; HZ, hypertrophic zone. (D) cKOosx mice showed an expansion of hypertrophic zone at P7 and P14, and the phenotype became normalized by P21. (E) No obvious difference was noted in the development of secondary ossification centers (arrow) between cKOosx and control tibiae at P14. (F) cKOosx mice showed enchondroma-like lesions (arrow) in the bone marrow at P7 and P14 . Right panels show higher magnification. Scale bars at the right bottom of each panel indicate 100μm.
To totally inactivate ERK1 and ERK2 in hypertrophic chondrocytes, we crossed ERK1−/−; ERK2flox/flox mice with Osx-Cre; ERK1−/−; ERK2+/flox mice. Osx-Cre; ERK1−/−; ERK2flox/flox mice (cKOosx) were born at the expected Mendelian ratio. The cKOosx mice were grossly normal in size at birth, but these mice showed a growth delay postnatally. At 3 weeks of age, cKOosx mice weighed only about one-third of ERK1−/−; ERK2flox/flox mice and half of Osx-Cre; ERK1−/−; ERK2+/flox mice (Fig.1A, Supplemental Fig 1D). The long bones of cKOosx mice were significantly shorter than those of control littermates (Fig 1B, Supplemental Fig.1A, B). The cKOosx mice invariably died around 3 weeks of age, which may be due to Osx-Cre expression in other systems(18).
cKOosx mice exhibit delayed formation of primary ossification centers and expansion of the zone of hypertrophic chondrocytes
Histological analysis of the tibia and femur demonstrated that the zone of hypertrophic chondrocytes was consistently wider in the growth plate of cKOosx mice after E15.5. At E15.5, there was no obvious difference in the total bone length and relative proportion of the zone of hypertrophic chondrocytes, indicating that the initial transition from proliferative to hypertrophic chondrocytes is seemingly unaffected in cKOosx mice (Fig.1C). At E16.5, when the bone marrow cavity was fully developed in control mice, cKOosx mice showed a persistent presence of hypertrophic chondrocytes in the middle of the diaphysis, indicating a delay in the formation of primary ossification centers. While the zone of hypertrophic chondrocytes remained expanded in cKOosx mice from E17.5 through P14, this phenotype gradually normalized after birth, and no difference was noted between cKOosx and control littermate mice at 3 weeks of age (Fig.1C, D). Despite the delay in the formation of primary ossification centers, cartilage mineralization was not affected in cKOosx mice (Supplemental Fig.2A). In addition, chondrocyte proliferation was not affected in cKOosx mice (Supplemental Fig.2B). Histological analysis of 2-week-old mice showed that there was no obvious difference in the development of the secondary ossification centers between cKOosx and control littermate mice (Fig.1E). The postnatal bone phenotype of cKOosx mice will be further characterized and described elsewhere. Another interesting phenotype in the long bones of cKOosx mice was cartilaginous islands resembling enchondroma in the bone marrow. The enchondroma-like lesions began to form at about P2 and persisted at least up to 3 weeks of age (Fig.1F, Supplemental Fig.1C, 1E, and data not shown). Histological analysis at P3 showed abundant cartilage remnants resembling cartilaginous islands in the bone marrow, and these cartilaginous remnants are still connected to the growth plate (Supplemental Fig.1E). These observations suggest that the cartilaginous islands develop from the unresorbed cartilage.
Expansion of the hypertrophic zone in cKOosx mice is caused by conditional deletion of ERK1 and ERK2 in hypertrophic chondrocytes but not in osteoblasts
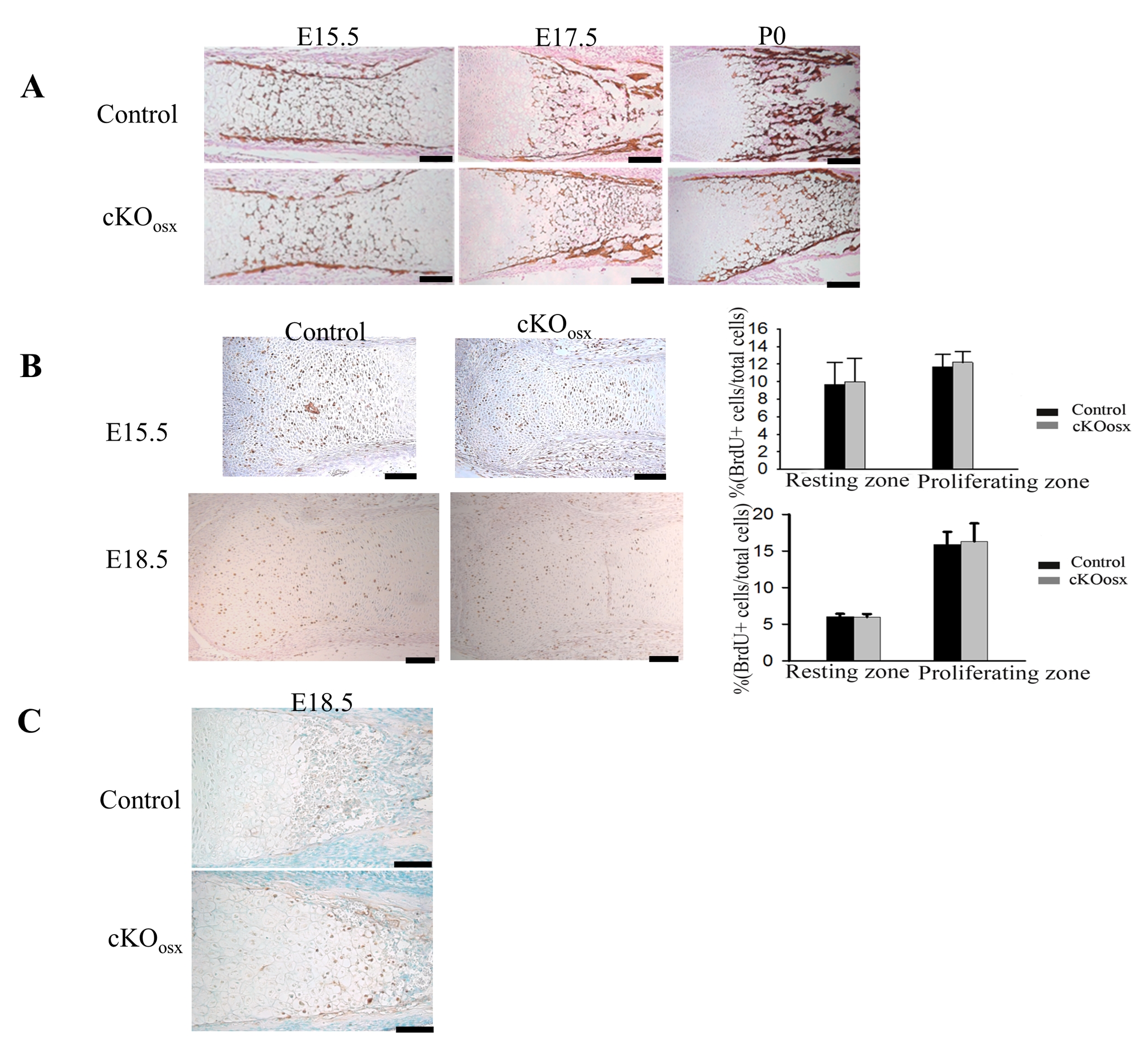
Osx-Cre mice have been shown to display Cre recombinase activity both in osteoblasts and hypertrophic chondrocytes in the growth plate(7,8). Consistent with previous reports, we observed X-gal staining in hypertrophic chondrocytes as well as cells in the osteoblast lineage in mice harboring the Osx-Cre transgene and the ROSA26-LacZ reporter allele (Fig.2B). We also examined the inactivation of ERK1/2 in cKOosx mice by immunohistochemistry. While chondrocytes in the resting and proliferating zones showed intense staining in cKOosx mice, staining for ERK1/2 was remarkably reduced in hypertrophic chondrocytes (Fig.2A). These observations suggest that the expansion of the zone of hypertrophic chondrocytes in cKOosx mice is caused by ERK1/2 inactivation in hypertrophic chondrocytes. We further inactivated ERK1/2 in osteoblasts by using the Col1a1CreER-DsRed transgene that does not show Cre recombinase activity in chondrocytes(9). We found no significant difference in the width of the zone of hypertrophic chondrocytes in Col1a1CreER-DsRed; ERK1−/−; ERK2flox/flox mice compared to controls at E18.5, even though ERK2 was efficiently deleted from osteoblasts in these animals (Fig. 2C). These results indicate that ERK1/2 deletion in hypertrophic chondrocytes but not in osteoblasts leads to the expansion of the zone of hypertrophic chondrocytes in cKOosx mice.
Figure 2. Conditional deletion of ERK1/2 in hypertrophic chondrocytes, but not in osteoblasts, causes expansion of the hypertrophic zone.

(A) Immunostaining for ERK1/2 of cKOosx and control tibiae at E15.5. Boxed areas (a-d) are magnified in the corresponding panel. While immunoreactivity for ERK1/2 is reduced in hypertrophic chondrocytes of cKOosx mice (b), intense staining is observed in hypertrophic chondrocytes of control mice (a) as well as in chondrocytes in the proliferating (c) and resting (d) zones of cKOosx mice. (B) X-gal staining of the tibia of Osx-Cre; ROSA26-LacZ mice at P0 demonstrated Cre recombinase activity in hypertrophic chondrocytes in addition to cells in the osteoblast lineage. (C) Alcian blue staining of Col1a1CreER-DsRed; ERK1−/−; ERK2flox/flox and control tibiae at E18.5. Tamoxifen was injected intraperitoneally into the pregnant mother at E13.5, E14.5 and E15.5, and embryos were harvested for histology at E18.5. The inactivation of ERK2 in the tibial diaphysis was about 80% as determined by quantitative real-time PCR. ERK1/2 deletion in osteoblasts does not cause an expansion of the hypertrophic zone. Data represent mean ± standard error, n=3 per group. Scale bars at the right bottom of each panel indicate 100μm.
Impaired chondrocyte terminal differentiation in cKOosx mice
The expansion of the zone of hypertrophic chondrocytes could be the result of decreased apoptosis, reduced vascular invasion, impaired cartilage resorption, accelerated hypertrophic chondrocyte differentiation, and delayed terminal chondrocyte differentiation. We therefore analyzed chondrocyte apoptosis by TUNEL staining. We found that there was an increase in the number of TUNEL-positive hypertrophic chondrocytes in cKOosx mice (Supplemental Fig.2C). Since increased apoptosis is expected to result in an opposite phenotype, i.e. the reduction of the zone of hypertrophic chondrocytes, apoptosis is unlikely to account for the expansion of the zone of hypertrophic chondrocytes in cKOosx mice.
The expansion of the zone of hypertrophic chondrocytes may occur through delayed cartilage resorption as a result of impaired vascular invasion. Therefore, we examined localization of vascular endothelial cells by immunostaining for a specific marker, von Willebrand factor (vWF). At E16.5, immunoreactivity for vWF was restricted to the diaphyseal bone collar in cKOosx mice, correlating with the delayed formation of primary ossification centers in these mice (Supplemental Fig. 3A). However, at E18.5, when the primary ossification center was formed in cKOosx mice, the pattern of vWF immunostaining at the chondro-osseous junction was indistinguishable between cKOosx mice and control littermates, indicating no major differences in vascular recruitment at this stage (Supplemental Fig. 3B). We also examined Vegf, an essential regulator of vascular invasion, by in situ hybridization. Vegf expression in hypertrophic chondrocytes was comparable between cKOosx mice and control littermate mice at E18.5 (Supplemental Fig.4A). These data suggest that impaired angiogenesis is unlikely to be the cause for the expansion of the zone of hypertrophic chondrocytes in cKOosx mice.
Impaired osteoclastogenesis can also lead to the expansion of the zone of hypertrophic chondrocytes. We assessed the number of osteoclasts at the chondro-osseous junction by tartrate-resistant acid phosphatase (TRAP) staining at E18.5 and P7. We found no differences in the number of TRAP positive cells at the chondro-osseous junction between cKOosx and control littermate mice (Supplemental Fig.3D, 3E). Moreover, immunostaining of Mmp9, a matrix metalloprotease that is highly expressed in osteoclasts/chondroclasts, showed no obvious difference at the chondro-osseous junction between cKOosx and control littermates at E18.5 (Supplemental Fig.3C). We also examined Rankl and Opg, genes involved in osteoclast differentiation, by quantitative real-time PCR. There was no significant difference in Rankl, Opg expression and Rankl/Opg ratio between cKOosx mice and control littermates at E15.5 (Fig.3). Taken together, these observations suggest that the expansion of the zone of hypertrophic chondrocytes is not caused by decreased osteoclast differentiation and their recruitment to the chondro-osseous junction.
Figure 3. Decreased expression of chondrocyte terminal differentiation markers in cKOosx mice.

Quantitative real-time PCR analysis of the tibia of ERK1−/−; ERK2flox/flox (WT-like), Osx-Cre; ERK1−/−; ERK2+/flox (CON), and Osx-Cre; ERK1−/−; ERK2flox/flox (cKO) embryos at E15.5. The inactivation of ERK2 in the E15.5 tibiae was about 30% in cKOosx mice compared with ERK1−/−; ERK2flox/flox mice, reflecting ERK2 inactivation that was restricted to hypertrophic chondrocytes. Osteopontin, Mmp13, Egr1, and Egr2 were significantly downregulated in cKOosx mice. Data represent mean ± standard error, n=5 per group. An asterisk (*) denotes a statistically significant difference compared with control group, p≤0.05.
Accelerated hypertrophic differentiation of chondrocytes from the proliferative to the early hypertrophic stage and delayed terminal differentiation of chondrocytes from the early hypertrophic stage to the terminal stage, could also account for the expansion of the zone of hypertrophic chondrocytes. In order to address these possibilities, we analyzed stage-specific chondrocyte markers Col2a1 (resting and proliferating chondrocytes), Ihh (prehypertrophic chondrocytes), and Col10a1 (hypertrophic chondrocytes) in the tibia by in situ hybridization at E15.5-E18.5. At all stages examined, cKOosx tibiae showed a normal pattern of Col2a1 and Ihh expression (Fig.4A-C). In contrast, the expression domain of Col10a1 was expanded at all stages, correlating with the expansion of the zone of hypertrophic chondrocytes. Furthermore, the area of terminally differentiated chondrocytes, where hypertrophic chondrocytes are past the Col10a1-expressing stage and express Osteopontin and Mmp13, was also expanded in the tibiae of cKOosx mice (Fig.4C), indicating that the expansion of the zone of hypertrophic chondrocytes was for the most part caused by an increase in the number of terminally differentiated chondrocytes. In addition, Osteopontin and Mmp13 expression in the terminally differentiated chondrocytes was dramatically reduced in cKOosx mice (Fig.4A-C), suggesting impaired differentiation of early hypertrophic chondrocytes toward terminally differentiated chondrocytes. Reduced expression of Osteopontin and Mmp13 and normal expression of Col10a1 in cKOosx cartilage was also confirmed by real-time PCR analysis of tibiae at E15.5 (Fig.3, data not shown). Collectively, these results indicate that in the endochondral bone of cKOosx mice, the terminal differentiation process of early hypertrophic chondrocytes was severely impaired, while chondrocyte differentiation from the proliferative stage to the early hypertrophic stage was unaffected. The impaired terminal differentiation most likely accounts for the expansion of the zone of hypertrophic chondrocytes in cKOosx mice.
Figure 4. In situ hybridization of chondrocyte markers Col2a1, Col10a1, Ihh, Osteopontin, and Mmp13.

Expression of terminal differentiation markers Osteopontin and Mmp13 was significantly reduced in the tibiae of cKOosx mice at E15.5 (A), E16.5 (B), and E18.5 (C), while Col2a1, Col10a1, and Ihh were expressed at similar levels compared with control. In figure C, the yellow dashes indicate the locations of the chondro-osseous junction, while the red dashes indicate the upper border of Osteopontin and Mmp13 expression domains. Scale bars at the right bottom of each panel indicate 100μm.
ERK1/2 inactivation in hypertrophic chondrocytes likely delays chondrocyte terminal differentiation in part through transcriptional factors Egr1 and Egr2
To identify downstream targets of ERK1/2 in hypertrophic chondrocytes that control terminal differentiation, we examined several candidate transcriptional factors by in situ hybridization and real-time PCR. We found no differences in Runx2 and Sox9 expression between cKOosx and control littermates at E15.5 and E18.5 (Fig. 3, Supplement Fig.4B, and data not shown). We also examined immediate early genes such as Egr1 and Egr2 that have been implicated in skeletal development(19). Real-time PCR analysis indicated that Egr1 and Egr2 were strongly downregulated in the tibiae of cKOosx embryos, and Nab2, a co-repressor of Egr1 and Egr2, was also slightly decreased in these embryos (Fig. 3). Interestingly, immunohistochemical analysis indicated that the expression of both Egr1 and Egr2 was restricted to hypertrophic chondrocytes adjacent to the chondro-osseous junction (Fig.5A), indicating co-expression with Osteopontin and Mmp13. Consistent with the real-time PCR results, Egr1 and Egr2 protein levels were strikingly reduced in cKOosx mice compared with control littermates (Fig.5A).
Figure 5. Regulation of ERK1/2 on chondrocyte terminal differentiation was partially through transcription factors Egr1/2.

(A) Immunostaining for Egr1 and Egr2 in E18.5 cKOosx and control tibiae. Color was developed using brown substrate for Egr1 and blue substrate for Egr2. Boxed areas (1-2) are magnified in the corresponding panels. Scale bars at the right bottom of each panel indicate 100μm. (B) Egr1, Egr2, and constitutively active MEK1 activate human Osteopontin promoter in vitro. Rat chondrosarcoma cells were co-transfected with OPN(−1206)-luc construct and expression plasmids for Egr1, Egr2, Nab2, and constitutively active MEK1. The promoter activity of cells transfected with pcDNA3.1 empty vectors was designated as 1. Data represent mean ± standard error, n=3 per group. An asterisk (*) denotes a statistically significant difference in the promoter activity compared with the pcDNA3.1 group, P<0.05. A pound sign (#) denotes a statistically significant difference in the promoter activity between MEK1 expression alone and MEK1 and Nab2 co-expression, P<0.05. (C) Proposed model for the effect of ERK1/2 on chondrocyte terminal differentiation. ERK1/2 regulates chondrocyte terminal differentiation at least in part through Egr1 and Egr2. ERK1/2 inactivation in hypertrophic chondrocytes results in impaired terminal differentiation and the formation of enchondroma-like lesions in the bone marrow.
To further examine the role of ERK MAPK signaling and Egr1 and Egr2 in terminal chondrocyte differentiation, we tested their effects on the promoter activity of Osteopontin in RCS cells, a chondrocyte cell line derived from rat chondrosarcoma(16). Expression of Egr1 and Egr2 increased the activity of OPN(−1206)-luc, a luciferase reporter construct harboring a 1.2 kb human Osteopontin promoter, similar to the expression of a constitutively active mutant of MEK1. Furthermore, co-expression of Nab2, a co-repressor of Egr1 and Egr2, strongly inhibited Egr1 and Egr2-induced and MEK1-induced activation of the Osteopontin promoter, suggesting that the activation of Osteopontin promoter by MEK1 signaling is at least in part mediated by Egr1 and Egr2 (Fig.5B and data not shown). Similar results were also obtained with the −914/+5 OPN-luc construct harboring a mouse Osteopontin promoter (data not shown). These observations support a model, in which MEK1-ERK signaling promotes Osteopontin expression in part through Egr1 and Egr2 (Fig.5C).
Discussion
During endochondral ossification, chondrocytes undergo sequential steps of differentiation leading to chondrocyte hypertrophy, in which chondrocytes lose Col2a1 expression and initiate Col10a1 expression. Hypertrophic chondrocytes further switch their transcriptional programs and undergo terminal differentiation prior to cell death through apoptosis(1). During this transition, chondrocytes lose Col10a1 expression and upregulate a new set of genes, including matrix metalloproteinase-13 (Mmp13) and Osteopontin. The precise mechanisms of how this terminal differentiation process takes place remain largely unknown. In the present study, we inactivated ERK1 and ERK2 in hypertrophic chondrocytes using the Osx-Cre transgene, while maintaining ERK MAPK signaling in chondrocytes in the resting and proliferating zones. The study allowed precise assessment of terminal hypertrophic chondrocyte differentiation by excluding the effects of ERK inactivation on earlier differentiation processes. Our results showed that the loss of ERK1 and ERK2 in hypertrophic chondrocytes severely impaired chondrocyte terminal differentiation, indicating that ERK MAPK signaling is essential for chondrocyte terminal differentiation. These observations are consistent with recently published in vitro data(20).
Since various cytokines and growth factors activate the ERK MAPK pathway, and ERK MAPK plays a crucial role in terminal differentiation, it is critically important to identify upstream signals that control ERK activation in hypertrophic chondrocytes. One possible regulator of ERK activity in hypertrophic chondrocytes is Fgfr1 signaling. Fgfr1 is specifically expressed in hypertrophic chondrocytes in the growth plate, and its deletion from chondrocytes results in the expansion of the zone of hypertrophic chondrocytes in conjunction with reduced Osteopontin expression (21), mimicking the phenotype of cKOosx mice. Other studies have also implicated EGFR signaling in chondrocyte terminal differentiation. EGFR is one of the strong activators of the ERK MAPK pathway. Consistent with the notion that EGFR signaling regulates ERK activity in hypertrophic chondrocytes, mice deficient in Egfr(22)and mice deficient in molecules involved in Egfr activation—TGFα(23) and ADAM17(24)—show an expansion of the zone of hypertrophic chondrocytes. These mice display reduced expression of Mmp13(22-24) similar to our cKOosx mice. Interestingly, Mmp13-null mice also show a transient expansion of the zone of hypertrophic chondrocytes(25,26). These observations are consistent with our finding that Mmp13 is a downstream target of the ERK MAPK pathway in hypertrophic chondrocytes.
Since hypertrophic chondrocytes switch their transcriptional programs when the cells undergo terminal differentiation, the transcriptional mechanisms whereby ERK signaling promotes terminal differentiation are of considerable interest. One possible mediator of ERK signaling is Runx2(27), a transcription factor that is essential for osteoblast differentiation and hypertrophic chondrocyte differentiation. Indeed, Runx2 has been shown to regulate Osteopontin and Mmp13 gene expression in osteoblasts(28,29). However, despite severe impairment of chondrocyte terminal differentiation in cKOosx mice, Runx2 was normally expressed in these mice. Although Runx2 activity may be regulated by ERK at the posttranslational level(30), downstream targets Osterix, Vegf, Ihh, and Col10a1 are normally expressed in cKOosx mice. These observations suggest that Runx2 is transcriptionally active even in the absence of ERK signaling, and therefore, Runx2 is unlikely to account for the abnormal growth plate phenotype of cKOosx mice.
In our present study, we identified transcription factors Egr1 and Egr2 as potential downstream targets of ERK1/2 in chondrocyte terminal differentiation. Interestingly, Egr1 and Egr2 expression was restricted to the last layers of hypertrophic chondrocytes, corresponding to the expression domain of Osteopontin and Mmp13, suggesting their roles in terminal differentiation. Our transient transfection experiments suggested that Egr1 and Egr2 mediate the activation of Osteopontin promoter by MEK-MAPK signaling in chondrocytes. Consistent with our observations, Egr1 has been shown to bind to the Osteopontin promoter in vascular smooth muscle cells(31). Genetic studies in mice have also implicated Egr1 and Egr2 in the regulation of terminal chondrocyte differentiation(19,32). Taken together, our observations suggest that MEK-MAPK signaling regulates chondrocyte terminal differentiation at least in part through Egr1 and Egr2.
Another interesting phenotype in the long bones of cKOosx mice is enchondroma-like lesions in the bone marrow. These lesions are reminiscent of human metachondromatosis, a human skeletal disorder caused by heterozygous mutations in the PTPN11 gene(33). PTPN11 encodes a tyrosine phosphatase SHP2 that promotes ERK MAPK signaling(34). Postnatal inactivation of Ptpn11 in mice results in the development of metachondromatosis-like exostoses and enchondromas that are characterized by reduced ERK signaling(35,36). Therefore, our observations in cKOosx mice provide strong evidence for the role of reduced ERK signaling in the development of enchondromas in Ptpn11 mutant mice and in human metachondromatosis. Defective terminal differentiation caused by the reduced activity of ERK may account for the development of enchondromas in Ptpn11 mutant mice and in metachondromatosis patients.
There are several weaknesses in the current study. First, we cannot totally rule out the possibility that ERK1/2 inactivation outside of hypertrophic chondrocytes also contributes to the aberrant skeletal growth of cKOosx mice. In this mouse model, ERK1/2 is also inactivated in osteoblasts. In addition, premature lethality of cKOosx mice might be caused by ERK1/2 inactivation elsewhere. The conditional inactivation of ERK1/2 specifically in hypertrophic chondrocytes would be helpful in addressing these possibilities. Second, given the roles of ERK1/2 in lineage specification of osteochondro progenitor cells(4), aberrant differentiation of osteogenic cells might account for the development of the enchondroma-like lesions in the bone marrow. Third, we were not able to assess the skeletal phenotype in older mice due to their premature lethality. Transient inactivation of the Osx-Cre transgene by doxycycline treatment may allow investigation in older mice and help us determine their similarities to human metachondromatosis.
In summary, our results demonstrate that ERK1 and ERK2 in hypertrophic chondrocytes are essential regulators of chondrocyte terminal differentiation. Transcription factors Egr1 and Egr2 may be involved in this process. The proposed model for the roles of ERK1 and ERK2 in chondrocyte terminal differentiation is summarized in Figure 5C. During normal long bone development, ERK1 and ERK2 act as positive regulators for the transition of early hypertrophic chondrocytes to terminally differentiated chondrocytes, which express high levels of terminal differentiation markers, such as Osteopontin and Mmp13. These processes are at least in part mediated by Egr1 and Egr2. When ERK1 and ERK2 are deleted from hypertrophic chondrocytes, the expression of Egr1 and Egr2 is dramatically downregulated resulting in impaired chondrocyte terminal differentiation. The growth plate exhibits reduced expression of terminal differentiation markers Osteopontin and Mmp13, expansion of the zone of hypertrophic chondrocytes, and an overall delay in endochondral ossification. In postnatal development, enchondroma-like lesions develop in the bone marrow, presumably as a result of impaired terminal chondrocyte differentiation, while the expansion of the growth plate gradually normalizes. Since cKOosx mice display enchondroma-like lesions postnatally, this novel mouse model will serve as an excellent model for studying the pathogenesis of enchondroma. Further analyses will provide novel insights into the roles of the ERK MAPK pathway in skeletal development and human diseases.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Teresa Pizzuto for expert technical assistance and Valerie Schmedlen for editorial assistance.
This work was supported by National Institutes of Health grant R01AR055556 to S.M and E.M.G., and the Ray A. Kroc and Robert L. Kroc Scholars Award in Arthritis and Connective Tissue Diseases Research to S.M.
Footnotes
Authors’ roles: Study design: ZC, SM. Data collection: ZC, SXY. Data analysis and interpretation: ZC, SM. Drafting and revising manuscript: ZC, SM, GZ, EMG. Approving final manuscript: all authors. ZC and SM take responsibility for the integrity of the data analysis.
Disclosures All the authors state that they have no conflicts of interest.
Supplemental Figures 1-4 and Supplemental Table 1 are submitted as supplemental data.
References
- 1.Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol. 2013;5(1):a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26(22):3113–21. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 3.Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–69. doi: 10.1146/annurev-genom-091212-153523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol. 2009;29(21):5843–57. doi: 10.1128/MCB.01549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18(3):290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 7.Rankin EB, Wu C, Khatri R, Wilson TL, Andersen R, Araldi E, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell. 2012;149(1):63–74. doi: 10.1016/j.cell.2012.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ouyang Z, Chen Z, Ishikawa M, Yue X, Kawanami A, Leahy P, et al. Prx1 and 3.2kb Col1a1 promoters target distinct bone cell populations in transgenic mice. Bone. 2014;58:136–45. doi: 10.1016/j.bone.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bukulmez H, Balmes G, et al. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet. 2009;18(2):227–40. doi: 10.1093/hmg/ddn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senkel S, Lucas B, Klein-Hitpass L, Ryffel GU. Identification of target genes of the transcription factor HNF1beta and HNF1alpha in a human embryonic kidney cell line. Biochim Biophys Acta. 2005;1731(3):179–90. doi: 10.1016/j.bbaexp.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, de Belle I, Liang H, Adamson ED. Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Mol Cell. 2004;15(1):83–94. doi: 10.1016/j.molcel.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 13.Fatherazi S, Matsa-Dunn D, Foster BL, Rutherford RB, Somerman MJ, Presland RB. Phosphate regulates osteopontin gene transcription. J Dent Res. 2009;88(1):39–44. doi: 10.1177/0022034508328072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeBlanc SE, Jang SW, Ward RM, Wrabetz L, Svaren J. Direct regulation of myelin protein zero expression by the Egr2 transactivator. J Biol Chem. 2006;281(9):5453–60. doi: 10.1074/jbc.M512159200. [DOI] [PubMed] [Google Scholar]
- 15.Murakami S, Kan M, McKeehan WL, de Crombrugghe B. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 2000;97(3):1113–8. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhopadhyay K, Lefebvre V, Zhou G, Garofalo S, Kimura JH, de Crombrugghe B. Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-alpha 1(II) collagen gene. J Biol Chem. 1995;270(46):27711–9. doi: 10.1074/jbc.270.46.27711. [DOI] [PubMed] [Google Scholar]
- 17.Davey RA, Clarke MV, Sastra S, Skinner JP, Chiang C, Anderson PH, et al. Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res. 2012;21(4):885–93. doi: 10.1007/s11248-011-9581-z. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Shi Y, Regan J, Karuppaiah K, Ornitz DM, Long F. Osx-Cre Targets Multiple Cell Types besides Osteoblast Lineage in Postnatal Mice. PLoS One. 2014;9(1):e85161. doi: 10.1371/journal.pone.0085161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levi G, Topilko P, Schneider-Maunoury S, Lasagna M, Mantero S, Cancedda R, et al. Defective bone formation in Krox-20 mutant mice. Development. 1996;122(1):113–20. doi: 10.1242/dev.122.1.113. [DOI] [PubMed] [Google Scholar]
- 20.Bowen ME, Ayturk UM, Kurek KC, Yang W, Warman ML. SHP2 regulates chondrocyte terminal differentiation, growth plate architecture and skeletal cell fates. PLoS Genet. 2014;10(5):e1004364. doi: 10.1371/journal.pgen.1004364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacob AL, Smith C, Partanen J, Ornitz DM. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol. 2006;296(2):315–28. doi: 10.1016/j.ydbio.2006.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Siclari VA, Lan S, Zhu J, Koyama E, Dupuis HL, et al. The critical role of the epidermal growth factor receptor in endochondral ossification. J Bone Miner Res. 2011;26(11):2622–33. doi: 10.1002/jbmr.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Usmani SE, Pest MA, Kim G, Ohora SN, Qin L, Beier F. Transforming growth factor alpha controls the transition from hypertrophic cartilage to bone during endochondral bone growth. Bone. 2012;51(1):131–41. doi: 10.1016/j.bone.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Saito K, Horiuchi K, Kimura T, Mizuno S, Yoda M, Morioka H, et al. Conditional inactivation of TNFalpha-converting enzyme in chondrocytes results in an elongated growth plate and shorter long bones. PLoS One. 2013;8(1):e54853. doi: 10.1371/journal.pone.0054853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131(23):5883–95. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, et al. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci U S A. 2004;101(49):17192–7. doi: 10.1073/pnas.0407788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida CA, Furuichi T, Fujita T, Fukuyama R, Kanatani N, Kobayashi S, et al. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat Genet. 2002;32(4):633–8. doi: 10.1038/ng1015. [DOI] [PubMed] [Google Scholar]
- 28.Hess J, Porte D, Munz C, Angel P. AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J Biol Chem. 2001;276(23):20029–38. doi: 10.1074/jbc.M010601200. [DOI] [PubMed] [Google Scholar]
- 29.Sato M, Morii E, Komori T, Kawahata H, Sugimoto M, Terai K, et al. Transcriptional regulation of osteopontin gene in vivo by PEBP2alphaA/CBFA1 and ETS1 in the skeletal tissues. Oncogene. 1998;17(12):1517–25. doi: 10.1038/sj.onc.1202064. [DOI] [PubMed] [Google Scholar]
- 30.Xiao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, et al. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J Biol Chem. 2000;275(6):4453–9. doi: 10.1074/jbc.275.6.4453. [DOI] [PubMed] [Google Scholar]
- 31.Liu QF, Yu HW, Liu GN. Egr-1 upregulates OPN through direct binding to its promoter and OPN upregulates Egr-1 via the ERK pathway. Mol Cell Biochem. 2009;332(1-2):77–84. doi: 10.1007/s11010-009-0176-4. [DOI] [PubMed] [Google Scholar]
- 32.Le N, Nagarajan R, Wang JY, Svaren J, LaPash C, Araki T, et al. Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci. 2005;8(7):932–40. doi: 10.1038/nn1490. [DOI] [PubMed] [Google Scholar]
- 33.Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafe L, Superti-Furga A, et al. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet. 2011;7(4):e1002050. doi: 10.1371/journal.pgen.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grossmann KS, Rosario M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. 2010;106:53–89. doi: 10.1016/S0065-230X(10)06002-1. [DOI] [PubMed] [Google Scholar]
- 35.Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature. 2013;499(7459):491–5. doi: 10.1038/nature12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim HK, Feng GS, Chen D, King PD, Kamiya N. Targeted disruption of Shp2 in chondrocytes leads to metachondromatosis with multiple cartilaginous protrusions. J Bone Miner Res. 2014;29(3):761–9. doi: 10.1002/jbmr.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.