

Abstract
Background
Defects in programmed cell death, or apoptosis, are a hallmark of cancer. The anti-apoptotic B-cell lymphoma 2 (BCL-2) family proteins, including BCL-2, BCL-XL, and MCL-1 have been characterized as key survival factors in multiple cancer types. Because cancer types with BCL2 and MCL1 amplification are more prone to inhibition of their respectively encoded proteins, we hypothesized that cancers with a significant frequency of BCL2L1 amplification would have greater dependency on BCL-XL for survival.
Methods
To identify tumor subtypes that have significant frequency of BCL2L1 amplification, we performed data mining using The Cancer Genome Atlas (TCGA) database. We then assessed the dependency on BCL-XL in a panel of cell lines using a selective and potent BCL-XL inhibitor, A-1155463, and BCL2L1 siRNA. Mechanistic studies on the role of BCL-XL were further undertaken via a variety of genetic manipulations.
Results
We identified colorectal cancer as having the highest frequency of BCL2L1 amplification across all tumor types examined. Colorectal cancer cell lines with BCL2L1 copy number >3 were more sensitive to A-1155463. Consistently, cell lines with high expression of BCL-XL and NOXA, a pro-apoptotic protein that antagonizes MCL-1 activity were sensitive to A-1155463. Silencing the expression of BCL-XL via siRNA killed the cell lines that were sensitive to A-1155463 while having little effect on lines that were resistant. Furthermore, silencing the expression of MCL-1 in resistant cell lines conferred sensitivity to A-1155463, whereas silencing NOXA abrogated sensitivity.
Conclusions
This work demonstrates the utility of characterizing frequent genomic alterations to identify cancer survival genes. In addition, these studies demonstrate the utility of the highly potent and selective compound A-1155463 for investigating the role of BCL-XL in mediating the survival of specific tumor types, and indicate that BCL-XL inhibition could be an effective treatment for colorectal tumors with high BCL-XL and NOXA expression.
Keywords: BCL-XL, MCL-1, NOXA, BCL-XL inhibitor, Colorectal cancer
Background
Cancer is a genetic disease that arises from the accumulation of somatic gene alterations. One way to identify key genes in cancer is to examine genomic regions that undergo frequent alterations. Recent technological advances in characterizing these alterations make it possible to identify genes that are essential for the initiation and/or survival of cancer, and thus encode potential therapeutic targets.
Evasion of apoptosis is a hallmark of cancer cells. One mechanism of apoptotic pathway deregulation is via upregulation of the anti-apoptotic BCL-2 family members. Apoptotic pathway proteins belong to a family of BCL-2 Homology (BH)-domain-containing proteins comprising three classes: 1) multi-domain anti-apoptotic (BCL-2, BCL-XL, BCL-W, BFL-1/A1, and MCL-1), 2) multi-domain pro-apoptotic (BAX, BAK), and 3) BH3-only pro-apoptotic (BID, BIM, BAD, BIK, NOXA, PUMA, BMF, and HRK). The BH3-only proteins contain a single BH3 domain and are bound by specific anti-apoptotic proteins [1]. For example, BCL-2 and BCL-XL bind and antagonize BIM but not NOXA. In contrast, MCL-1 and A1 bind and antagonize NOXA but not BAD protein. Other BH3 domain proteins such as BIM and PUMA are bound and antagonized by all anti-apoptotic proteins. BAX and BAK are known as the “effectors”. Once activated, these proteins oligomerize on the outer mitochondrial membrane and induce pore formation; this results in the release of cytochrome c and other pro-apoptotic proteins that ultimately carry out the cell death mechanism.
The role of anti-apoptotic BCL-2 family proteins in various cancers has been well studied [2]. BCL-2 was initially identified from the breakpoint of the t(14;18) chromosomal translocation found in over 60 % of indolent B cell non-Hodgkin’s lymphoma [3–6]. In addition to the vast majority of follicular lymphomas, many germinal center B cell (GCB) subtype diffuse large B cell lymphomas (DLBCL) also exhibit the t(14;18) chromosomal translocation [7–9]. BCL2 amplification is also detected in many hematologic malignancies such as the activated B cell-like (ABC) subtype of DLBCL [10]. Not surprisingly, cell lines with the translocation or amplification are more sensitive to the selective BCL-2 inhibitor ABT-199 [11].
MCL1 was reported to be amplified in 10.9 % of tumor samples analyzed, spanning multiple cancer subtypes [12]. Fluorescence in situ hybridization (FISH) of the MCL1 region identified lung and breast cancers as having significantly higher frequencies of focal amplification, suggesting that these tumors depend on MCL-1 for survival. This is supported by multiple studies demonstrating that cell lines with MCL1 amplification are sensitive to siRNA knockdown of MCL1 [12, 13].
BCL-XL has been implicated as a key survival factor in numerous solid tumors [2]. Based on the evidence that cancer types with BCL2 and MCL1 amplification are more prone to inhibition of their encoded proteins, we hypothesized that cancers with a significant frequency of BCL2L1 amplification are more dependent on BCL-XL for survival. In this study, we identified colorectal cancer as having a significant incidence of BCL2L1 amplification. We then dissected the role of BCL-XL in colorectal cancer cell lines using a selective small-molecule inhibitor of BCL-XL and a variety of genetic manipulations.
Materials and methods
Reagents
BCL-XL inhibitor A-1155463 and navitoclax were synthesized at AbbVie, Inc. (North Chicago, IL). All the siRNAs were purchased from Dharmacon (Lafayette, CO).
Cell culture, transfection, and cell-based assays
Colorectal cell lines (ATCC) were cultured in RPMI (Invitrogen, Carlsbad, CA) supplemented with 10 % fetal bovine serum (FBS) (Invitrogen), 1 % sodium pyruvate (Invitrogen), and 4.5 g/L glucose (Sigma, MO), or DMEM (Invitrogen) supplemented with 10 % FBS. All the lines were maintained in a humidified chamber at 37 °C containing 5 % CO2.
LS1034, SW1417, GEO, and RKO cells were transfected in 6-well plates with siRNAs using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). A final concentration of 20 nM siRNA was used in all cases. The sense sequences of the BCL-XL siRNA used is ACAAGGAGAUGCAGGUAUUUU (Dharmacon). The sense sequences of the MCL-1 siRNAs used is GCATCGAACCATTAGCAGATT (Dharmacon). The cells were then grown in medium without antibiotic before harvesting for western blotting analysis. LS1034 cells were transfected at 1.5–2.5 × 104 cells/100 μl in 96-well tissue culture plates with 20 nM Noxa siRNA pool (Dharmacon). The cells were grown in medium without antibiotic before harvesting.
Cells were treated with increasing concentration of A-1155463. Cells were assayed for viability after 72 h using the CellTiter-Glo luminescent cell viability assay according to the manufacturer’s protocol (Promega, Madison, WI). Results were normalized to cells without treatment. EC50 was calculated using the GraphPad Prism software (La Jolla, CA).
Western blot analysis
Cell lysates were prepared in RIPA buffer (Sigma) plus protease inhibitor cocktail (Roche). 20 μg of total protein was resolved on a 12 % SDS polyacrylamide gel and probed with anti-BCL-XL (Epitomics, Burlingame, CA), anti-MCL-1 (Epitomics), anti-BCL-2 (BD), anti-BIM (Epitomics), anti-actin and anti-NOXA (Abcam, Cambridge, MA). Antibody against tubulin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used as a loading control.
Fluorescence-activated cell sorting (FACS) analysis
LS1034 cells were treated with DMSO or 200 nM A-1155463, with or without 50 μM Z-VAD caspase inhibitor (Santa Cruz Biotechnology, Inc.) for 72 h. DNA content was measured by flow cytometry to determine the effect of the inhibitors on the cell cycle and cell death. Following treatment, cells were spun down, the medium was removed and the cells were resuspended in staining solution (50 μg propidium iodide, 40 U/ml RNase, 0.1 % triton X-100 in PBS) at a cell concentration of 1 × 106 cells/ml. The cells were stored in the dark at room temperature for at least 30 min or up to 24 h at 4 °C before the DNA content was determined by flow cytometry using a FACS Calibur (BD Biosciences, San Jose, CA).
Results
BCL-XL has been implicated as a key survival factor in numerous solid tumors. Since cancer types with BCL2 and MCL1 amplification are more prone to inhibition of these proteins, we reasoned that significant frequency of BCL2L1 amplification in a particular cancer type may indicate dependency on BCL-XL for survival. To identify cancer types with a significant amplification of BCL2L1, we utilized The cBio Cancer Genomics Portal (http://cbioportal.org), an open-access resource for interactive exploration of multidimensional cancer genomics data sets from more than 5,000 tumor samples from 20 cancer studies [14]. We surveyed the percentage of copy number alternations of BCL2, BCL2L1 and MCL1 among a majority of these tumor samples (Fig. 1a) and found that colorectal cancer has the highest percentage of BCL2L1 gain/amplification among all cancer types. Out of 212 samples, 15 % have BCL2L1 amplification, while MCL1 amplification is found in only 1 %. BCL2 amplifications were not observed, although homozygous deletion is observed in 1 % of colorectal cancer samples (data not shown). Furthermore, there is a good correlation between BCL2L1 copy number gain/amplification and gene expression as determined by RNAseq and microarray analysis (Fig. 1b and data not shown). Cervical cancer also has a high percentage of BCL2L1 gains/amplifications (Fig. 1a) although the sample numbers are limited. Consistent with other studies, we found that DLBCL has the highest percentage of BCL2 amplification (11 %) and lung adenocarcinoma has the highest percentage of MCL1 amplification (20 %) (Fig. 1a). These data indicated that each tumor type has a selective dependency on a particular anti-apoptotic protein for survival and colorectal and cervical tumors are potentially more dependent on BCL-XL for survival.
Fig. 1.
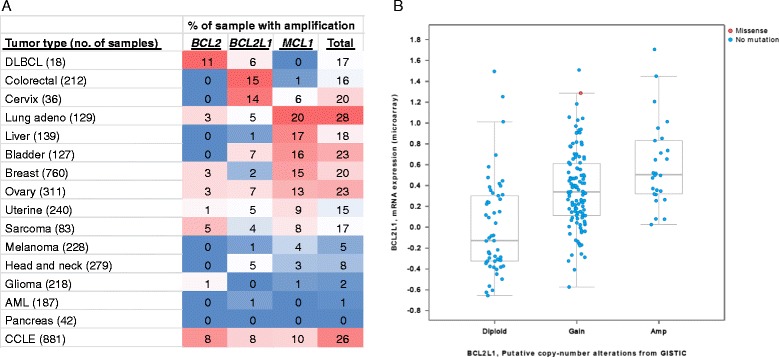
Cross-cancer tumor sample analysis identified colorectal cancer as having the highest frequency of BCL2L1 amplification. a Cross-cancer alteration summary for BCL2, BCL2L1 and MCL1 based on data obtained from cBioPortal for Cancer Genomics. b Correlation between gene expression and copy number alteration of BCL2L1 in 212 colorectal cancer samples
To test this hypothesis, we selected a panel of 25 colorectal cancer cell lines with known copy number of BCL2L1. We first investigated the sensitivity of these colorectal cell lines to treatment with the selective BCL-XL inhibitor, A-1155463 ([15] and Fig. 2a). This recently reported small molecule is more potent in inhibiting BCL-XL than navitoclax but shows negligible activity against BCL-2 or MCL-1, thus making it an excellent tool for dissecting BCL-XL cancer biology. A-1155463 demonstrated strong growth inhibition of over half of the colorectal cell lines (Fig. 2b) as defined by EC50 values ≤0.5 μM in the presence of 10 % FBS. Consistent with its lower potency against BCL-XL, navitoclax induced cell death in the same cell lines, although at higher concentrations (data not shown).
Fig. 2.
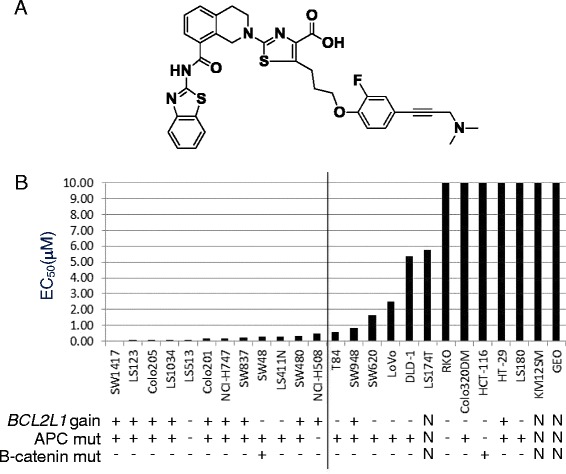
Colon cancer cell lines with BCL2L1 gain are more sensitive to BCL-XL inhibitor. a Structure of the potent and specific BCL-XL inhibitor A-1155463. b Colorectal cell lines were treated with increasing concentrations of A-1155463. Cells were assayed for viability after 72 h. Results were normalized to cells without treatment. Each cell line was treated with A-1155463 in at least 3 different experiments and the average EC50 is presented. BCL2L1 copy number gain >3 is indicated with + or -. Mutation status of APC and β-catenin is also indicated with + or -. These data are taken from the CCLE database [24]. N denotes cases where data were not available
Previously, we and others have shown that copy number (CN) gain, protein and mRNA expression of BCL-2 family members is a crucial determinant of sensitivity to BH3 mimetics such as ABT-737, an inhibitor of BCL-2, BCL-XL, and BCL-W, and ABT-199, a specific inhibitor of BCL-2 [11, 16–18]. Herein we report for the first time that cell lines with BCL2L1 gains are sensitive to selective BCL-XL inhibition. Among the 11 lines with BCL2L1 gain (CN > 3), 9 are sensitive to A-1155463 (EC50 < 0.5 μM). In contrast, only 3 out of 11 lines with no CN gain of BCL2L1 are sensitive to A-1155463 (Fig. 2b).
To determine if expression of BCL-2 family members plays a role in the sensitivity of colorectal cell lines to A-1155463, we collected lysates from these cell lines and performed western blotting analysis. Colorectal cell lines within our panel expressed varying levels of MCL-1 and BCL-XL (Fig. 3a). In general, there was an inverse relationship between the expression of MCL-1 versus NOXA and BCL-XL. We observed a strong correlation (0.68) between cell lines with BCL2L1 gain and BCL-XL protein expression. In addition, sensitive lines had higher protein expression of BCL-XL (p = 0.0002) and NOXA (p = 0.02), higher BCL-XL to MCL-1 ratio (p = 0.0027), and a trend towards lower expression of MCL-1, although not statistical significant (p = 0.12). The Pearson correlation between log EC50 and expression of BCL-XL, NOXA, MCL-1, and BCL-XL/MCL-1 was 0.75, 0.653, 0.363, and 0.637, respectively. The Spearman correlation between EC50 and expression of BCL-XL, NOXA, MCL-1, and BCL-XL/MCL-1 was 0.789, 0.749, 0.277, and 0.795, respectively. BCL-2 expression was detected in a single line, consistent with other studies showing that BCL-2 expression is less common in solid tumor cell lines [19]. To confirm these findings, we performed correlation analyses with mRNA expression data. There was a strong correlation between protein and mRNA expression for MCL-1 and BCL-XL. Additionally, sensitive lines had higher expression of BCL2L1 (p = 0.0078) and NOXA (p = 0.02), a higher BCL2L1 to MCL1 ratio (p = 0.0086), and a trend towards lower expression of MCL1 mRNA, although not statistical significant (p = 0.15) (Fig. 3b). The correlation coefficients for BCL2L1 and MCL1 versus sensitivity were −0.41 and 0.56, respectively, while the correlation coefficient for the BCL2L1 to MCL1 ratio was −0.56. These data indicate that BCL2L1 and NOXA expression may play important roles in determining sensitivity to selective BCL-XL inhibitors.
Fig. 3.
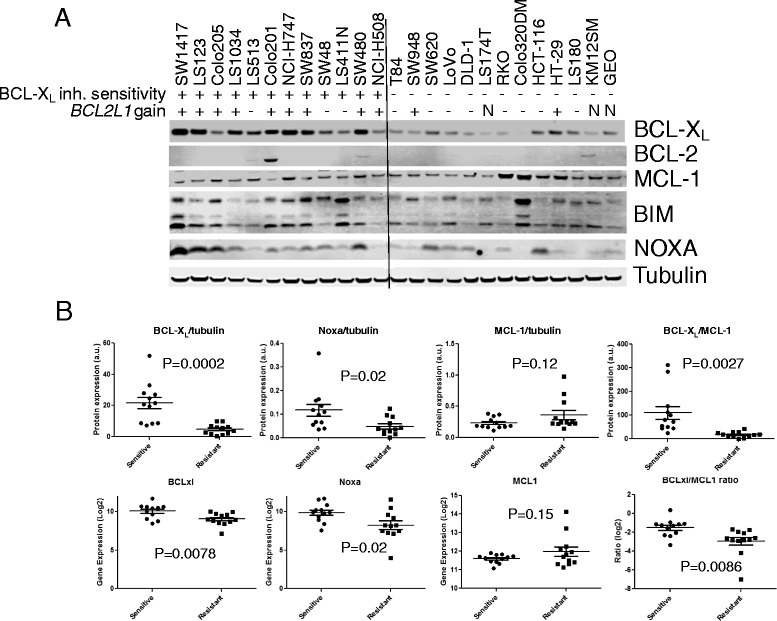
Colon cancer cell lines with high BCL-XL and NOXA are more sensitive to BCL-XL inhibitor. a Protein expression of BCL-2 family members in colorectal cancer cell lines. BCL2L1 copy number gains >3 is indicated with + or -. b Protein and mRNA expression of BCL-XL, NOXA, MCL-1, and ratio of BCL-XL /MCL-1 in A-1155463-sensitive vs. resistant colorectal cancer cell lines
Two recent manuscripts reported that BCL-XL is regulated by the β-catenin pathway. In an unbiased screen of 242 genomically characterized cancer cell lines with an Informer Set of 354 small molecules, cell lines with β-catenin mutation were reported to be amongst the most sensitive to navitoclax (ABT-263), an inhibitor of BCL-2, BCL-XL and BCL-W [20]. In a separate study, β-catenin-active cancers were reported to be dependent on a signaling pathway involving the transcriptional regulator YAP1 and the transcription factor TBX5, both of which form a complex with β-catenin [21]. This complex was further shown to be important for regulating BCL-XL and the anti-apoptotic gene BIRC5. APC is part of a destruction complex that mediates the ubiquitin-targeted proteolysis of β-catenin. Mutations in APC destabilize this complex and lead to the accumulation of β-catenin, which can then translocate to the nucleus and aberrantly activate the transcription of key drivers of oncogenesis [22]. Interestingly, a small subgroup of colorectal tumors with wild-type APC has point mutations in β-catenin that allow it to escape degradation [23]. Since BCL-XL was described to be regulated by the β-catenin pathway and multiple components of the pathway are highly mutated in colorectal cancer, we determined whether a correlation exists between these mutations and the activity of inhibitor A-1155463 in the colorectal cancer cell lines. While the majority of these lines have mutations in APC, only two lines have β-catenin mutations according to the Cancer Cell Line Encyclopedia (CCLE) [24]. Contrary to recent reports, we found no correlation between APC or β-catenin mutation status and dependence on BCL-XL for survival as revealed by sensitivity to A-1155463 (Fig. 2b) or navitoclax (data not shown). This could be explained by differences in cell lines between the studies. In particular, our studies focus on colon cell lines only. It is also possible that the disparity results from the lack of appropriately selective small molecules in the study by Basu et al. [20].
To confirm the survival dependence on BCL-XL within the colorectal cell line panel, we silenced its expression via small interfering RNA (siRNA) oligonucleotides specific to BCL2L1 in two sensitive (LS1034 and SW1417) and two resistant (RKO and GEO) colorectal cell lines. The two sensitive lines also exhibit BCL2L1 gain. BCL2L1 siRNA effectively knocked down BCL-XL protein expression completely in three cell lines, and ~80 % in SW1417 cells (Fig. 4a). However, the viability of only the A-1155463-sensitive colorectal cell lines was greatly reduced after transfecting BCL2L1 siRNA (defined as >50 % reduction). In contrast, there was minimal effect on the viability of the inhibitor-resistant cell lines, thus confirming that BCL-XL is critical for survival of a subgroup of colorectal cell lines. The viability is also confirmed using another assay that quantifies the ATP present in culture as an indicator of metabolically active cells (Fig. 4b). We found that the viability measured with this method is similar to that as shown in Fig. 4a, confirming that these A-1155463-sensitive colorectal cell lines indeed depend on BCL-XL for survival.
Fig. 4.
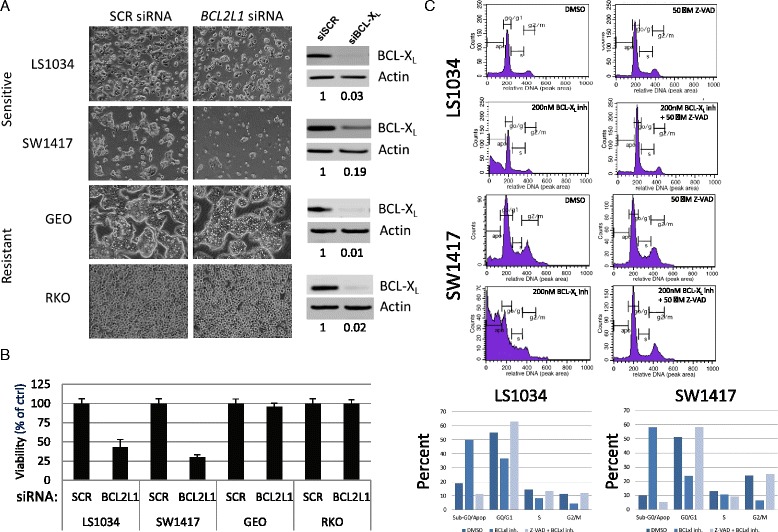
A-1155463 sensitivity correlates with the effect of BCL-XL knockdown and apoptosis. BCL2L1 or control siRNA was transfected into two A-1155463-sensitive cell lines (LS1034 and SW1417) and two resistant cell lines (GEO and RKO). a Viability was determined after 72 h by microscopy. Western blotting showing the degree of BCL-XL knock-down by BCL2L1 siRNAs. Levels of BCL-XL knockdown were measured by densitometry and shown as BCL-XL/actin. b Viability was determined after 72 h with CellTiter Glo reagent. c BCL-XL inhibitor induces apoptosis through activation of caspase-3/7 pathway. LS1034 and SW1417 cells were treated with DMSO or 200 nM A-1155463, with or without 50 μM Z-VAD caspase inhibitor for 24 h. DNA content was measured by flow cytometry to determine the effect of the inhibitors on the cell cycle and cell death. The percentage of cells in each phase of the cell cycle was plotted
To assess whether the BCL-XL inhibitor induced cell death in sensitive colorectal cell lines via apoptosis, we evaluated the cell cycle distribution of LS1034 and SW1417 cells after treatment with A-1155463 in the presence or absence of the caspase-3/7 inhibitor Z-VAD. As shown in Fig. 4c, A-1155463 treatment led to substantial accumulation of sub-G0/apoptosis cells (>50 %). This effect was completely abrogated in the presence of Z-VAD, indicating that cell killing occurs via the activation of caspase-3/7 pathway.
We investigated the role of MCL-1 in determining sensitivity to the BCL-XL inhibitor in two resistant colorectal cell lines (RKO and GEO) by silencing the expression of MCL-1 with siRNA. While silencing MCL-1 alone had no effect (data not shown), MCL-1 knockdown in the presence of A-1155463 afforded complete cell killing (Fig. 5a). Similar effects were observed with an additional resistant line, HT-29 (data not shown). These data indicate that MCL-1 plays a role in modulating sensitivity to BCL-XL inhibitors in colorectal cancer cell lines.
Fig. 5.
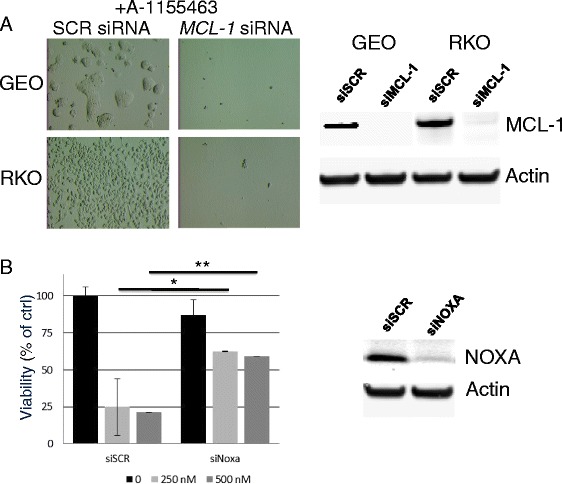
Silencing MCL1 sensitizes colorectal cells to BCL-XL inhibitor while silencing NOXA renders resistance to BCL-XL inhibitor. a MCL1 or control siRNA were transfected into RKO and GEO cells in the presence of A-1155463. Viability was determined after 48 h by microscopy. Western blotting showing the degree of MCL-1 knockdown by MCL1 siRNAs in GEO and RKO cells. b NOXA or control siRNA was transfected into LS1034 cells in the presence or absence of A-1155463. Viability was determined after 48 h. Results were normalized to cells transfected with scrambled siRNA control and presented as mean +/− SD (n = 3). *p < 0.01, **p < 0.005. Western blotting showing the degree of NOXA knockdown by NOXA siRNA pool
NOXA is a pro-apoptotic protein that antagonizes MCL-1 activity. Because NOXA expression is higher in the sensitive lines (Fig. 3a), we sought to assess its role in determining sensitivity to BCL-XL inhibitors. We thus asked whether silencing NOXA would rescue sensitive colorectal lines from treatment with A-1155463. Transfection of LS1034 cells with NOXA siRNA rendered the cells insensitive to A-1155463 treatment (Fig. 5b), indicating that NOXA function, likely involving its inhibition of MCL-1, is required to sensitize this cell line to BCL-XL inhibition. Similar results were observed in another BCL-XL inhibitor-sensitive line, SW1417 (data not shown).
Discussion
In this study, we set out to first identify cancer types that show high levels of BCL2L1 amplification using genomic mining. Our second major aim was to investigate the dependency of corresponding cell lines on BCL-XL for survival. To this end, we used not only siRNA but a recently disclosed small molecule inhibitor of BCL-XL that shows high potency and selectivity against other pro-survival proteins. Our study identified colorectal and cervical cancers as having the most frequent BCL2L1 amplification. Further studies showed that the majority of colorectal cancer cell lines with a gain in BCL2L1 are sensitive to the selective BCL-XL inhibitor A-1155463 as well as to siRNA-medited knockdown of BCL2L1. Although the gains are typically modest (3–4.4 copies), it should be noted that CN gain of other anti-apoptotic genes such as MCL1 is also modest in other tumors that nevertheless become dependent on these genes for survival [12]. In addition, the sensitive lines exhibit higher protein and mRNA expression of BCL-XL and NOXA, and a trend towards lower protein and mRNA expression of MCL-1 than the resistant lines.
Despite a recent report that navitoclax-sensitive cell lines are enriched with β-catenin mutations [20], we found no correlation between BCL-XL dependence and the presence of β-catenin or APC mutations. Possible reasons for this discrepancy could be differences in the colorectal cell lines used in the two studies, or alternatively, the fact that the highly selective BCL-XL inhibitor was not available at the time of the earlier study by Basu et al. and the study was thus confounded by the mixed selectivity of the compound used [20]. Our data is consistent with those of Rosenbluh et al., who reported that β-catenin-active cancers regulate BCL-XL as a survival factor [21]. In this regard, we found that sensitive lines are enriched with an active β-catenin pathway as determined by a β-catenin transcriptional reporter assay. Although this is a small sample size, preliminary data show that five out of six of the sensitive lines display active β-catenin pathway versus three out of six in the resistant group (data not shown). Collectively, our data indicate that BCL-XL may be regulated through multiple pathways in colorectal cancer, including CN gain and active β-catenin signaling. The role of other pathways cannot be ruled out.
Similar to navitoclax, we found that MCL-1 is a resistance factor for the BCL-XL-selective inhibitor A-1155463. Mechanistically, A-1155463 and navitoclax displace pro-apoptotic protein BIM from BCL-XL, which leads to the activation of apoptosis [2, 25]. In resistant cells, it is likely that the BIM released by BCL-XL inhibitor is readily sequestered by MCL-1, thereby inhibiting the activation of the apoptotic pathway. Indeed, it has been shown that MCL-1 and BCL-XL play compensatory roles in regulating apoptosis in most solid tumor cell lines [13, 16, 26]. Our experiments further demonstrate that silencing MCL-1 can sensitize solid tumor cell lines to selective BCL-XL inhibition, expanding on earlier studies with the BCL-2/BCL-XL inhibitors ABT-737 and ABT-263 [27–30]. By targeting MCL-1 via mTOR inhibition, colorectal cancers with Kras or BRAF mutations are sensitized to BCL-2/BCL-XL inhibition [31]. Our observation that silencing NOXA could rescue sensitive cells from A-1155463 also supports the role of MCL-1 as a resistance factor. NOXA is known to interact with MCL-1 and neutralize its activity by targeting this protein for degradation [1]. Thus, silencing NOXA decreases the antagonism of MCL-1, leaving it free to sequester pro-apoptotic proteins and prevent the activation of apoptosis [13].
Other gene amplifications have served as therapeutic targets for other cancers. For example, the MET and ALK tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung and gastric cancer according to MET and ALK alterations [32]. By utilizing genomic data to identify tumors with BCL2L1 gain, we identified colorectal cancers as being potentially more dependent on BCL-XL and more susceptible to BCL-XL inhibition. Indeed, by using a potent and selective BCL-XL inhibitor, A-1155643, we have shown that BCL-XL inhibition is sufficient to kill colon cancer cell lines expressing high levels of BCL-XL and the MCL-1-neutralizing protein NOXA. Of particular interest, sensitive lines are enriched in a gain of BCL2L1, suggesting BCL2L1 gain could be used as a patient stratification biomarker. This finding demonstrates the power of identifying potential new targets by examining genomic regions that undergo frequent alterations as well as the utility of highly potent and selective tool molecules. Finally, BCL-XL selective inhibitors may have utility as therapeutics for the treatment of colorectal cancer, a disease for which there is significant unmet medical need.
Conclusions
BCL-XL inhibition could be an effective treatment for colorectal tumors with high BCL-XL and NOXA expression. Additionally, BCL2L1 gain may represent stratification biomarker for colorectal cancer patients undergoing treatment with a BCL-XL inhibitor. Future studies should investigate the utility of BCL2L1 gain as a stratification biomarker.
Acknowledgements
We thank the AbbVie Oncology Biomarkers group for discussion and critical review of the manuscript.
Footnotes
Competing interests
All authors are employees of AbbVie. The design, study conduct, and financial support for this research was provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the manuscript.
Authors’ contributions
HZ: Designed and performed experiments. JX: Performed immunoblotting. PH: Performed experiments. SKT: Performed FACS. JC: Performed experiments. SJ: Performed experiments. AJS: Conceived experiments and wrote paper. JDL: Conceived experiments and wrote paper. LTL: Conceived, designed, performed experiments and wrote paper. All authors read and approved the final manuscript.
Contributor Information
Haichao Zhang, Email: Haichao.zhang@abbvie.com.
John Xue, Email: John.xue@abbvie.com.
Paul Hessler, Email: Paul.hessler@abbvie.com.
Stephen K. Tahir, Email: Stephen.k.tahir@abbvie.com
Jun Chen, Email: Jun.chen@abbvie.com.
Sha Jin, Email: Sha.jin@abbvie.com.
Andrew J. Souers, Email: Andrew.souers@abbvie.com
Joel D. Leverson, Email: Joel.leverson@abbvie.com
Lloyd T. Lam, Email: Lloyd.Lam@abbvie.com
References
- 1.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 2.Lam LT, Zhang H, Chyla B. Biomarkers of therapeutic response to BCL2 antagonists in cancer. Mol Diagn Ther. 2012;16:347–56. doi: 10.1007/s40291-012-0003-6. [DOI] [PubMed] [Google Scholar]
- 3.Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/S0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 4.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 5.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–3. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 6.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229:1390–3. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- 7.Iqbal J, Sanger WG, Horsman DE, Rosenwald A, Pickering DL, Dave B, et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am J Pathol. 2004;165:159–66. doi: 10.1016/S0002-9440(10)63284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leich E, Salaverria I, Bea S, Zettl A, Wright G, Moreno V, et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood. 2009;114:826–34. doi: 10.1182/blood-2009-01-198580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang JZ, Sanger WG, Greiner TC, Staudt LM, Weisenburger DD, Pickering DL, et al. The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood. 2002;99:2285–90. doi: 10.1182/blood.V99.7.2285. [DOI] [PubMed] [Google Scholar]
- 10.Iqbal J, Neppalli VT, Wright G, Dave BJ, Horsman DE, Rosenwald A, et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol. 2006;24:961–8. doi: 10.1200/JCO.2005.03.4264. [DOI] [PubMed] [Google Scholar]
- 11.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 12.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, Guttikonda S, Roberts L, Uziel T, Semizarov D, Elmore SW, et al. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene. 2011;30:1963–8. doi: 10.1038/onc.2010.559. [DOI] [PubMed] [Google Scholar]
- 14.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett. 2014;5:1088–93. doi: 10.1021/ml5001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–83. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 17.Olejniczak ET, Van Sant C, Anderson MG, Wang G, Tahir SK, Sauter G, et al. Integrative genomic analysis of small-cell lung carcinoma reveals correlates of sensitivity to bcl-2 antagonists and uncovers novel chromosomal gains. Mol Cancer Res. 2007;5:331–9. doi: 10.1158/1541-7786.MCR-06-0367. [DOI] [PubMed] [Google Scholar]
- 18.Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maiga S, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood. 2011;118:3901–10. doi: 10.1182/blood-2010-11-317438. [DOI] [PubMed] [Google Scholar]
- 19.Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1 doi: 10.1038/cddis.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–61. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457–73. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neufeld KL, Zhang F, Cullen BR, White RL. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000;1:519–23. doi: 10.1093/embo-reports/kvd117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 24.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 26.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Jin S, Abraham V, Huang X, Liu B, Mitten MJ, et al. The Bcl-2/Bcl-XL/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol Cancer Ther. 2011;10:2340–9. doi: 10.1158/1535-7163.MCT-11-0415. [DOI] [PubMed] [Google Scholar]
- 28.Lam LT, Lu X, Zhang H, Lesniewski R, Rosenberg S, Semizarov D. A microRNA screen to identify modulators of sensitivity to BCL2 inhibitor ABT-263 (navitoclax) Mol Cancer Ther. 2010;9:2943–50. doi: 10.1158/1535-7163.MCT-10-0427. [DOI] [PubMed] [Google Scholar]
- 29.Lam LT, Roberts-Rapp L. Multiplex analysis of anti-apoptotic BCL2 family and caspase 3 activation by microbead arrays. Assay Drug Dev Technol. 2014;12:190–6. doi: 10.1089/adt.2013.557. [DOI] [PubMed] [Google Scholar]
- 30.Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA, et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26:3972–9. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 31.Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014;4:42–52. doi: 10.1158/2159-8290.CD-13-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J, Ou SH. Towards the goal of personalized medicine in gastric cancer–time to move beyond HER2 inhibition. Part II: Targeting gene mutations and gene amplifications and the angiogenesis pathway. Discov Med. 2013;16:7–14. [PubMed] [Google Scholar]