

Abstract
FCR, the standard of care for frontline treatment of CLL patients, is associated with a high rate of neutropenia and infectious complications. GM-CSF reduces myelosuppression and can potentiate rituximab activity. We conducted a clinical trial combining GM-CSF with FCR for frontline treatment of 60 CLL patients. Eighty-six percent completed all 6 courses and 18% discontinued GM-CSF for toxicity; grade 3–4 neutropenia was observed in 30% of cycles, and severe infections in 16% of cases. ORR was 100%. Both median EFS and OS have not been reached. Longer EFS was associated with favorable cytogenetic. GM-CSF led to a lower frequency of infectious complications than the historical FCR group, albeit similar EFS and OS.
Keywords: CLL, FCR, GM-CSF, neutropenia, infections
INTRODUCTION
Chronic Lymphocytic Leukemia (CLL) is the most common leukemia in western countries, with an incidence of 5/100.000 cases per year1. Its clinical course is highly variable and different prognostic criteria have been developed to predict outcome2.
CLL treatment has evolved from single-agent alkylator use, to purine nucleoside analogs, to their use in combination, and eventual addition of monoclonal antibodies3. A single institution phase II study suggested that fludarabine, cyclophosphamide and rituximab (FCR) improved outcome in patients with untreated CLL4, 5. The superiority of FCR over combination therapy with fludarabine and cyclophosphamide alone in the frontline therapy of patients with CLL has more recently been confirmed in a large randomized phase III trial thus making the FCR regimen standard of care in this setting6, 7.
In an effort to improve those results further, various modifications of FCR have been evaluated combining FCR with other agents such as alemtuzumab or mitoxantrone. None of those combinations however demonstrated any significant advance8, 9.
CLL cells express lower levels of surface CD20 than do most of the other B cell malignancies, likely explaining the inferior activity of rituximab in this disease. Granulocyte Macrophage Colony-Stimulating Factor (GM-CSF) may increase in vitro CD20 surface expression in CLL cells, although contrasting data are presented in literature10, 11. GM-CSF also enhances in vitro antibody-dependent cellular cytotoxicity of rituximab against CLL cells12. Preliminary data from combinations of rituximab with GM-CSF have been encouraging, both in terms of efficacy and safety13, although the addition of GM-CSF to other regimens and in more unfavorable clinical settings has not proved to be equally beneficial14, 15. Furthermore, GM-CSF has the potential advantage of reducing myelosuppression and thus infectious complications related to use of FCR.
We therefore designed a clinical study combining FCR with GM-CSF for previously untreated patients with CLL who met indications for therapy. Primary objectives included analysis of response and survival, as well as assessment of the rates of myelosuppression and infectious complications. We compared those results with a historical control of FCR-only treated patients from our database.
METHODS
Eligibility
Patients with untreated CLL (except for prior immunotherapy only), who met NCI-WG criteria for initiation of therapy16 were eligible. Accrual was limited to patients with beta-2-microglobulin (B2M) levels of less than twice the upper limit of normal (< 4 mg/L)(only 1 patient with a B2M of 4.2 mg/L was included). Alternative studies were in place at time of accrual for patients with higher B2M levels. Patients were excluded for ECOG performance status of 3 or more, active hepatitis B or congestive heart failure (NYHA of 3 or more and/or cardiac ejection fraction lower than 40%). All patients signed informed consent according to institutional guidelines. The study was approved by the Institutional Review Board of the University of Texas MD Anderson Cancer Center and was conducted in accordance with the basic principles of the Declaration of Helsinki. Accrual period was from September 2006 to June 2008 and follow up period was until September 2012. All patients were treated at MD Anderson Cancer Center (MDACC).
Dose and Schedule
Fludarabine 25 mg/m2 and cyclophosphamide 250 mg/m2 were administered intravenously on days 2–4 of course 1 and on days 1–3 of courses 2–6. Rituximab 375 mg/m2 intravenously was given on day 1 and of course 1 and 500 mg/m2 on day 1 during courses 2–6. GM-CSF 250 mcg/m2 was administered subcutaneously on days −1 and 5–11 of course 1 and on days −1 and 4–10 of courses 2–6. Prior to rituximab, patients received oral acetaminophen 650 mg and diphenhydramine 25–50 mg. Before chemotherapy, patients received ondansetron 24 mg intravenously. Patients also received prophylaxis for Pneumocystis jiroveci (carinii) pneumonia with trimethoprim/sulfamethoxazole or equivalent and antiviral prophylaxis with valacyclovir or equivalent for the duration of therapy. Courses were administered every 4–6 weeks as permitted by neutrophil and platelet recovery and/or resolution of toxicities (defined according to Common Toxicity Criteria 3). Patients who achieved a stable partial response or who had a continued response after the first 3 courses could receive up to a total of 6 courses. Patients with no response or progressive disease after 1–3 courses were considered as having failed therapy and were taken off treatment and observed for progression and survival.
Monitoring and Response Assessment
Monitoring of patients consisted of complete blood count (CBC) with differential every 1–2 weeks as long as on therapy and during 6 monthly follow-up visits thereafter as long as on study. Physical examination and serum chemistries including creatinine, transaminases and total bilirubin were repeated before each treatment course.
Patients were evaluated for response 30 days after the last course of treatment, according to NCI-WG criteria16, with physical examination, CBC with differential and bone marrow biopsy and aspiration.
Statistical Analysis
Event Free Survival (EFS) was defined as time from start of treatment to relapse or death. Overall Survival (OS) was defined as time from start of treatment to death or last follow up. Survival distributions were calculated using the method of Kaplan and Meier, and univariate comparisons were made using the log-rank test. Categorical and continuous variables were compared using the χ2 or Fisher exact tests, and the Mann-Whitney test, as appropriate. All P values were 2-sided and considered significant if equal to or less than 0.05.
RESULTS
Patient Characteristics
Patient characteristics are summarized in Table I. A total of 60 patients were enrolled.
Table I.
Patient characteristics (N=60)
| FCR+GMCSF (N=60) | FCR (N=166) | p | |
|---|---|---|---|
| MALES (n) | 44 (73%) | 118 (71%) | 0.74 |
|
| |||
| AGE (Y) | 55 (35–77) | 55 (17–86) | 0.92 |
|
| |||
| RAI III–IV | 13 (22%) | 42 (25%) | 0.57 |
|
| |||
| WBC (K/UL) | 78 (3–323) | 76 (7–619) | 1 |
|
| |||
| ALC (K/UL) | 71 (2–291) | 66 (3–557) | 1 |
|
| |||
| ANC (K/UL) | 3 (0–10) | 4 (0–32) | 0.55 |
|
| |||
| HB (g/dL) | 13.3 (8.1–16.4) | 13.5 (7.5–18) | 0.38 |
|
| |||
| PLT (K/UL) | 180 (35–420) | 185 (4–429) | 0.76 |
|
| |||
| B2M (mg/dL) | 2.8 (1.7–4.2) | 2.9 (1.6–3.9) | 0.24 |
|
| |||
| UNMUTATED (n) | 30/53 (57%) | 57/99 (57%) | 0.91 |
|
| |||
| ZAP70+ (n) | 28/53 (53%) | 34/67 (51%) | 0.82 |
|
| |||
| CD38 ≥ 30% (n) | 19/58 (33%) | 22/140 (16%) | 0.007 |
|
| |||
| FISH (n) | 59 | NA | NA |
| DEL13Q | 18 (30%) | ||
| NEG | 12 (20%) | ||
| +12 | 10 (17%) | ||
| DEL11Q | 17 (29%) | ||
| DEL17P | 2 (4%) | ||
Seventy-three percent of patients were males, median age was 55 (35–77) years and 22% had advanced stage disease (Rai III–IV). Median B2M was 2.8 (1.7–4.2) mg/L, 57% of patients had unmutated IGHV gene, 53% had positive ZAP70 by immunohistochemistry and 33% had positive CD38 by flow cytometry (≥30%). Fluorescence in situ hybridization (FISH) panel was available in 59 patients and showed deletion of 17p and 11q in 2 (4%) and 17 (29%) patients, respectively.
Treatment Characteristics and Toxicity
Patients received a median number of 6 courses (range 3–6) of FCR, with 49 (82%) patients receiving all 6 courses.
Eleven (18%) patients discontinued GM-CSF, after a median number of 3 (1–4) courses. Reasons for GM-CSF discontinuation were syncope-like reactions (2 cases), fever (1), pain/erythema at site of injection (4), patient choice (2), and diarrhea/vomiting (2). Three (5%) patients needed at least 1 dose reduction of FCR because of grade 3–4 toxicity.
Grade (G) 3–4 neutropenia was observed in 50 (83%) patients and in 103 of 340 (30%) cycles. Ten (17%) patients developed late neutropenia (defined as onset later than 3 months after the last cycle of therapy). G3–4 infectious complications were reported in 9 (16%) patients, including 1 fatal case (see Table II). One case of thrombocytopenia was immune-mediated. One death occurred on study, before completion of treatment. The patient died after 4 courses with a febrile syndrome, followed by respiratory failure and irreversible coma for several weeks; magnetic resonance imaging (MRI) of the brain showed hyper-densities without enhancement in the thalamic and cerebral pedunculus, with negative work-up for infectious etiologies of leukoencephalopathy.
Table II.
Toxicities
| Number (%) | |
|---|---|
| Hematological G3–4 | |
| Neutropenia | 50 (83%) |
| Anemia | 4 (7%) |
| Thrombocytopenia | 9 (15%) |
|
| |
| Infections G3–4 | |
| Pneumonia | 2 (4%) |
| FUO | 5 (8%) |
| Diarrhea | 1 (2%) |
| Encephalopathy | 1 (2%) |
Response and Survival
Complete remission (CR) was achieved in 45 (75%) patients, nodular partial remission (nPR) in 6 (15%) and PR in 9 (10%) patients, for an overall response rate of 100%. No differences in responses were observed according to prognostic factors (see Table III).
Table III.
Response according to prognostic factors (N=60)
| CR | nPR | PR | p | |
|---|---|---|---|---|
| MUTATED | 17 | 2 | 4 | 0.73 |
| UNMUTATED | 23 | 4 | 3 | |
|
| ||||
| CD38 < 30% | 31 | 3 | 7 | 0.46 |
| CD38 ≥ 30% | 14 | 3 | 2 | |
|
| ||||
| ZAP70− | 18 | 4 | 3 | 0.65 |
| ZAP70+ | 22 | 2 | 4 | |
|
| ||||
| DEL13Q | 12 | 2 | 4 | 0.56 |
| NEG | 10 | 0 | 2 | |
| +12 | 7 | 2 | 1 | |
| DEL11Q | 14 | 1 | 2 | |
| DEL17P | 1 | 1 | 0 | |
|
| ||||
| B2M<2 | 5 | 0 | 1 | 0.70 |
| B2M 2–3 | 24 | 2 | 4 | |
| B2M≥3 | 16 | 4 | 4 | |
|
| ||||
| RAI 0–II | 38 | 3 | 6 | 0.08 |
| RAI III–IV | 7 | 3 | 3 | |
With a median follow up of 56 (7–60) months, median EFS has not been reached and 17 (28%) patients have progressed. EFS was significantly longer in patients with favorable FISH abnormalities (p=0.005)(Figure 1). No significant differences in EFS were observed according to any other patient or treatment characteristics or degree of response.
Figure 1.
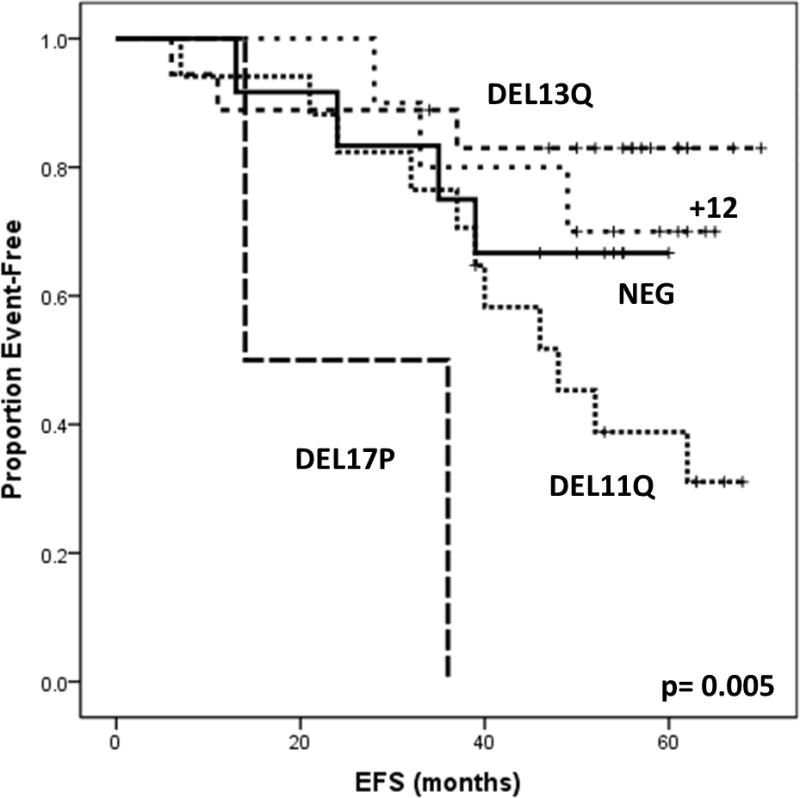
Event Free Survival (EFS) according to patient characteristics
Median OS has not been reached. No significant differences in OS were observed according to patient or treatment characteristics or degree of response. Nine patients died; 3 patients died of disease progression and 6 died in CR: 3 died of complications related to secondary myelodysplastic syndrome (MDS), 1 died of suicide, and 2 died of severe infections (1 during treatment). Secondary hematological malignancies were reported in 5 patients, including 4 cases of MDS, after a median time of 38 (35–39) months, and 1 case of acute lymphoblastic leukemia after 70 months.
Historical Comparison with FCR
We compared patients on study to a historical group of frontline CLL patients who were treated with FCR at our institution between 1999 and 2004. In order to minimize the bias deriving from selection criteria, the initial matching variable was B2M (less than 4 mg/L) and 166 patients were identified.
Other than a higher frequency of CD38 positivity in the study group (p=0.007), the 2 groups matched for all the remaining baseline characteristics (Table I). FISH testing was not routinely done at the time of the FCR accrual and it could not be included in the comparison. As several cycles were administered in the community setting, data regarding the use of granulocyte colony stimulating factor (G-CSF) were not available for comparison.
Patients on study had a higher rate of PR (p=0.03), and a lower incidence of G3–4 infections (p=0.05), when compared to the FCR group; the remaining treatment, response, and toxicity features did not differ significantly between the two groups. In particular, the incidence of secondary myeloid malignancies was not significantly increased in the study group (Table IV).
Table IV.
Historical comparison with FCR. (*) The rate of G3–4 neutropenia per cycle did not differ significantly between the 2 groups (30% vs 37%, p=0.30).
| Number (%) | FCR+GMCSF (N=60) | FCR (N=166) | p |
|---|---|---|---|
| 6 FCR cycles | 49 (82%) | 135 (81%) | 0.95 |
| ORR | 60 (100) | 162 (98%) | 0.58 |
| CR | 45 (75%) | 138 (83%) | 0.17 |
| nPR | 6 (15%) | 13 (8%) | 0.60 |
| PR | 9 (10%) | 10 (6%) | 0.03 |
| G3–4 neutropenia | 50 (83%) | 125 (75%) | 0.20 |
| G3–4 neutropenia (late) | 10 (17%) | 20/120 (17%) | 1 |
| G3–4 infections | 9 (15%) | 46 (28%) | 0.05 |
| Dose reduction | 3 (5%) | 17 (10%) | 0.29 |
| MDS | 4 (6%) | 5 (3%) | 0.25 |
The median follow up period was 56 (6–70) months for the study group and 121 (0–159) months for the FCR group. EFS and OS did not differ significantly between the two groups (p=0.21 and 0.46, respectively) (Figure 2).
Figure 2.
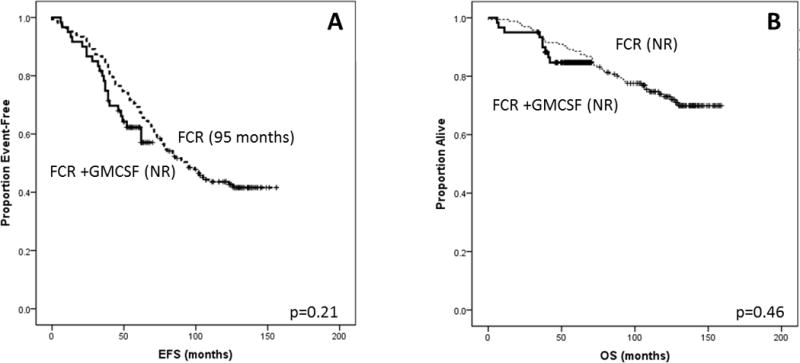
Historical comparison with FCR
DISCUSSION
FCR has become the standard of care for the frontline treatment of patients with CLL and a good performance status in the absence of significant comorbidities5, 6. FCR has also shown to be more effective in eradicating residual disease, which may account for the better outcome associated with its use17. However, FCR has been more frequently associated with grade 3 and 4 neutropenia and, in some patients, with prolonged myelosuppression6.
GM-CSF has shown to enhance the activity of rituximab thus possibly facilitating achievement of deeper responses10, 12, 13. Moreover, GM-CSF could mitigate the myelosuppressive effect of FCR and therefore allow achievement of adequate dose intensity on treatment. These observations have been corroborated by a recent retrospective analysis, showing improved Progression Free Survival (PFS) and OS in CLL patients treated with FCR in combination with growth factors18.
In our study, GM-CSF has been shown to be relatively safe and feasible when given in combination with FCR. The discontinuation rate was 18% and mostly related to local skin reactions. Of the 2 reported severe reactions, 1 was associated with an EDTA-containing formula, which was therefore changed. GM-CSF and G-CSF have comparable toxicity profile, albeit a higher rate of injection site reactions with the use of the former19. The absence of studies using comparable doses of G-CSF does not allow us to determine whether the reported discontinuation rate has to be considered high. However, the immunomodulatory role described for GM-CSF, in contrast to G-CSF20, and its potential benefit for disease control, not evident with the present follow-up, may support its use, although higher toxicity. The ORR was 100%, and was independent from baseline prognostic factors. Of note, the used selection criteria may have favorably biased the results, given the well described prognostic role of B2M levels21. A better EFS was described in association with favorable cytogenetics, as already demonstrated for FCR22–24. Two patients with deletion 17p (del17p) were included in the study. They achieved a CR and a nPR and their EFS was 36 and 14 months, respectively. Although the significance of these findings is limited by sample size, they may raise some interest, given the poor response to first-line treatment usually described for this cytogenetic subgroup25.
In order to assess the benefit provided by the addition of GM-CSF to FCR, we compared the study group to a historical group of patients in our database who received FCR only as frontline therapy. In order to minimize the bias deriving from selection criteria, the two groups were initially matched according to B2M levels, but showed later to be similar in all pretreatment characteristics. The only exception was CD38 expression. CD38 positive cells are more sensitive to CXCL12 signaling, with a higher propensity to home to lymphoid tissues, where they can more easily proliferate, clonally evolve and escape therapeutic agents26. Further molecular analyses are warranted to investigate any interaction between GM-CSF and CLL microenvironment. Of interest, no differences in EFS were observed between the 2 groups. Unfortunately, FISH analysis, another crucial prognostic factor, was not available for the historical group, limiting our comparison.
No differences in ORR were observed between the 2 groups. Sensitive methods of minimal residual disease detection (not available at our institution at time of both treatments) and longer follow up may address the question of whether or not the addition of GM-CSF to FCR plays a role also on disease-control. It still remains the question whether a safe and effective intensification of FCR might also be achieved adding biological drugs different from GM-CSF (such as lenalidomide or B cell receptor inhibitors) or by substituting the backbone FCR with other regimens (such as bendamustine and rituximab)27, 28. Ongoing and future trials might soon provide an answer to this therapeutic dilemma.
Although a comparable rate of neutropenia (both per patient and per cycle), the incidence of G3–4 infections was lower in the study group. When compared to data reported in the CLL8 trial, although a higher rate of G3–4 neutropenia (83 vs 34%) a lower rate of G3–4 infections was observed (15% vs 21%). Modulation of macrophage activity or neutrophil function may account for this phenomenon, although correlative studies are missing. Moreover, data about the use of G-CSF during the historical FCR study were not collected, limiting the comparison between the 2 groups.
No increase in the incidence of secondary myeloid neoplasms has been observed, as previously suspected for the co-administration of FCR and growth factors29. In our experience with frontline FCR, the risk of myelodysplasia at 6 years was 2.8%5. An incidence of 6% with a similar follow-up is obviously worrisome and may raise concern, albeit any definitive statement is limited by sample size.
Despite the lower frequency of severe infections, no benefit in OS was observed for the study group. Late infectious complications, unaffected by GM-CSF use30, could be speculated.
In conclusion, the addition of GM-CSF to FCR for frontline treatment of patients with CLL is feasible and not inferior to FCR alone. Its use may result in a decrease of the rate of early infectious complications, although caution should be used about the potential incidence of second myeloid malignancies.
Acknowledgments
None
Footnotes
Declaration of Interest: SF and AF received research support from Genetech.
References
- 1.Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol. 2007;139(5):809–19. doi: 10.1111/j.1365-2141.2007.06856.x. [DOI] [PubMed] [Google Scholar]
- 2.Mougalian SS, O’Brien S. Adverse prognostic features in chronic lymphocytic leukemia. Oncology (Williston Park) 2011;25(8):692–6. , 99. [PubMed] [Google Scholar]
- 3.Gribben JG, O’Brien S. Update on therapy of chronic lymphocytic leukemia. J Clin Oncol. 2011;29(5):544–50. doi: 10.1200/JCO.2010.32.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keating MJ, O’Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol. 2005;23(18):4079–88. doi: 10.1200/JCO.2005.12.051. [DOI] [PubMed] [Google Scholar]
- 5.Tam CS, O’Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112(4):975–80. doi: 10.1182/blood-2008-02-140582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–74. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 7.Casak SJ, Lemery SJ, Shen YL, et al. U.S. Food and drug administration approval: rituximab in combination with fludarabine and cyclophosphamide for the treatment of patients with chronic lymphocytic leukemia. Oncologist. 2011;16(1):97–104. doi: 10.1634/theoncologist.2010-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parikh SA, Keating MJ, O’Brien S, et al. Frontline chemoimmunotherapy with fludarabine, cyclophosphamide, alemtuzumab, and rituximab for high-risk chronic lymphocytic leukemia. Blood. 2011;118(8):2062–8. doi: 10.1182/blood-2011-01-329177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faderl S, Wierda W, O’Brien S, Ferrajoli A, Lerner S, Keating MJ. Fludarabine, cyclophosphamide, mitoxantrone plus rituximab (FCM-R) in frontline CLL <70 Years. Leuk Res. 2010;34(3):284–8. doi: 10.1016/j.leukres.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venugopal P, Sivaraman S, Huang XK, Nayini J, Gregory SA, Preisler HD. Effects of cytokines on CD20 antigen expression on tumor cells from patients with chronic lymphocytic leukemia. Leuk Res. 2000;24(5):411–5. doi: 10.1016/s0145-2126(99)00206-4. [DOI] [PubMed] [Google Scholar]
- 11.Yagci M, Akar I, Sucak GT, Haznedar R. GM-CSF does not increase CD20 antigen expression on chronic lymphocytic leukemia lymphocytes. Leuk Res. 2005;29(7):735–8. doi: 10.1016/j.leukres.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 12.Voso MT, Pantel G, Rutella S, et al. Rituximab reduces the number of peripheral blood B-cells in vitro mainly by effector cell-mediated mechanisms. Haematologica. 2002;87(9):918–25. [PubMed] [Google Scholar]
- 13.Ferrajoli A. Incorporating the use of GM-CSF in the treatment of chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50(3):514–6. doi: 10.1080/10428190902763541. [DOI] [PubMed] [Google Scholar]
- 14.Zent CS, Wu W, Bowen DA, et al. Addition of granulocyte macrophage colony stimulating factor does not improve response to early treatment of high-risk chronic lymphocytic leukemia with alemtuzumab and rituximab. Leuk Lymphoma. 2013;54(3):476–82. doi: 10.3109/10428194.2012.717276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsimberidou AM, O’Brien SM, Cortes JE, et al. Phase II study of fludarabine, cytarabine (Ara-C), cyclophosphamide, cisplatin and GM-CSF (FACPGM) in patients with Richter’s syndrome or refractory lymphoproliferative disorders. Leuk Lymphoma. 2002;43(4):767–72. doi: 10.1080/10428190290016872. [DOI] [PubMed] [Google Scholar]
- 16.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bottcher S, Ritgen M, Fischer K, et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol. 2012;30(9):980–8. doi: 10.1200/JCO.2011.36.9348. [DOI] [PubMed] [Google Scholar]
- 18.Gruber M, Fleiss K, Porpaczy E, et al. Prolonged progression-free survival in patients with chronic lymphocytic leukemia receiving granulocyte colony-stimulating factor during treatment with fludarabine, cyclophosphamide, and rituximab. Ann Hematol. 2011;90(10):1131–6. doi: 10.1007/s00277-011-1260-x. [DOI] [PubMed] [Google Scholar]
- 19.Bohlius J, Herbst C, Reiser M, Schwarzer G, Engert A. Granulopoiesis-stimulating factors to prevent adverse effects in the treatment of malignant lymphoma. Cochrane Database Syst Rev. 2008;(4):CD003189. doi: 10.1002/14651858.CD003189.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wierda WG, Kipps TJ. Gene therapy and active immune therapy of hematologic malignancies. Best Pract Res Clin Haematol. 2007;20(3):557–68. doi: 10.1016/j.beha.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Wierda WG, O’Brien S, Wang X, et al. Characteristics associated with important clinical end points in patients with chronic lymphocytic leukemia at initial treatment. J Clin Oncol. 2009;27(10):1637–43. doi: 10.1200/JCO.2008.18.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin KI, Tam CS, Keating MJ, et al. Relevance of the immunoglobulin VH somatic mutation status in patients with chronic lymphocytic leukemia treated with fludarabine, cyclophosphamide, and rituximab (FCR) or related chemoimmunotherapy regimens. Blood. 2009;113(14):3168–71. doi: 10.1182/blood-2008-10-184853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zenz T, Stilgenbauer S. Therapy with the FCR regimen does not overcome chronic lymphocytic leukemia biology: aberrant p53 expression predicts response and survival. Leuk Lymphoma. 2009;50(10):1559–61. doi: 10.1080/10428190903159384. [DOI] [PubMed] [Google Scholar]
- 24.Tsimberidou AM, Tam C, Abruzzo LV, et al. Chemoimmunotherapy may overcome the adverse prognostic significance of 11q deletion in previously untreated patients with chronic lymphocytic leukemia. Cancer. 2009;115(2):373–80. doi: 10.1002/cncr.23993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badoux XC, Keating MJ, Wierda WG. What is the best frontline therapy for patients with CLL and 17p deletion? Curr Hematol Malig Rep. 2011;6(1):36–46. doi: 10.1007/s11899-010-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. CD38 and chronic lymphocytic leukemia: a decade later. Blood. 2011;118(13):3470–8. doi: 10.1182/blood-2011-06-275610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smolej L. Modern concepts in the treatment of chronic lymphocytic leukemia. Hematology. 2009;14(5):249–54. doi: 10.1179/102453309X446153. [DOI] [PubMed] [Google Scholar]
- 28.Burger JA, Montserrat E. Coming full circle: 70 years of chronic lymphocytic leukemia cell redistribution, from glucocorticoids to inhibitors of B-cell receptor signaling. Blood. 2013;121(9):1501–9. doi: 10.1182/blood-2012-08-452607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y, Tang G, Medeiros LJ, et al. Therapy-related myeloid neoplasms following fludarabine, cyclophosphamide, and rituximab (FCR) treatment in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Mod Pathol. 2012;25(2):237–45. doi: 10.1038/modpathol.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tam C, Seymour JF, Brown M, et al. Early and late infectious consequences of adding rituximab to fludarabine and cyclophosphamide in patients with indolent lymphoid malignancies. Haematologica. 2005;90(5):700–2. [PubMed] [Google Scholar]