

Abstract
Large tumors exhibit high interstitial pressure heightened by growth against the constraining stroma. Such pressures could stimulate tumor proliferation via a mechanosensitive ion channel. We studied the effects of 0–80 mmHg increased extracellular pressure for 24 h on proliferation of SW620, Caco‐2, and CT‐26 colon; MCF‐7 breast; and MLL and PC3 prostate cancer cells, and delineated its mechanism in SW620 cells with specific inhibitors and siRNA. Finally, we compared NF‐kB, phospho‐IkB and cyclin D1 immunoreactivity in the high pressure centers and low pressure peripheries of human tumors. Pressure‐stimulated proliferation in all cells. Pressure‐driven SW620 proliferation required calcium influx via the T‐type Ca2+ channel Cav3.3, which stimulated PKC‐β to invoke the IKK‐IkB–NF–kB pathway to increase proliferation and S‐phase fraction. The mitotic index and immunoreactivity of NF‐kB, phospho‐IkB, and cyclin D1 in the center of 28 large human colon, lung, and head and neck tumors exceeded that in tumor peripheries. Extracellular pressure increases [Ca2+]i via Cav3.3, driving a PKC‐β‐ IKK‐ IkB–NF–kB pathway that stimulates cancer cell proliferation. Rapid proliferation in large stiff tumors may increase intratumoral pressure, activating this pathway to stimulate further proliferation in a feedback cycle that potentiates tumor growth. Targeting this pathway may inhibit proliferation in large unresectable tumors.
Keywords: Calcium channels, Cav3.3, PKC, NF‐kB, Pressure, Proliferation
Highlights
Increased extracellular pressure stimulates proliferation in diverse cancer cells.
Pressure induces Ca2+ influx through the T‐type Ca2+ channel Cav3.3.
Pressure‐induced Cav3.3 Ca2+ influx activates PKCβ and NF‐kB to promote proliferation.
The higher pressure centers of human tumors exhibit increased NF‐kB and IkB activity.
NF‐kB and IkB activity in tumors parallel cyclin D1 suggesting increased proliferation.
1. Introduction
Malignant tumor extracellular matrix is often stiffer than the matrix surrounding adjacent non‐malignant cells (Ingber, 2008). As solid tumors expand against constraining stroma, interstitial pressure increases by 4–50 mmHg relative to pressure within normal surrounding tissues (Gutmann et al., 1992; Less et al., 1992; Raju et al., 2008). Mathematical models (Sarntinoranont et al., 2003) and direct observation suggest higher pressures within large tumors' centers decrease toward their peripheries (Boucher et al., 1990). Such increased pressure impedes perfusion and delivery of chemotherapy to tumors (Navalitloha et al., 2006), but the direct effects of increased extracellular pressure on the tumor cells themselves are less clear.
Prolonged pressures similar to those in tumors stimulate proliferation in mesangial cells during glomerular hypertension, in cardiac myocytes after abdominal aortic constriction, and in endothelial cells (Bevan, 1976; Kawata et al., 1998; Schwartz et al., 1999). Our preliminary study found that 15 mmHg increased pressure stimulates SW620 and HCT‐116 colon cancer cell proliferation but did not define the mechanism of this effect (Walsh et al., 2004). Substrate stiffness and substrate deformation also influence cell growth in vitro (Kumar and Weaver, 2009; Paszek et al., 2005). This may occur through mechanosensitive ion channels, which influence processes ranging from bacterial turgor to growth in cardiac myocytes and epithelial cells (Hamill and Martinac, 2001).
Calcium is commonly transported by mechanosensitive ion channels and necessary for several cell processes (Hamill and Martinac, 2001). [Ca2+]i increases transiently in the G1/S transition of normal cells (Capiod et al., 2007) while sustained [Ca2+]i, due to T‐type channel over‐expression, causes androgen‐dependent LNCaP prostate cancer to assume a malignant apoptosis‐resistant neuroendocrine phenotype (Mariot et al., 2002). We sought to explore whether increased extracellular pressure stimulates proliferation in cancer cells by activating a mechanosensitive calcium channel. We then further investigated calcium‐sensitive mediators that modulate proliferation. This led us to the serine/threonine kinase PKC and the transcription factor NF‐kB. Our preliminary work suggested that mitogenic effects of pressure in colon cancer cells require PKC and are associated with PKCα membrane translocation (Walsh et al., 2004). NF‐kB modulates gene transcription in cell‐cycle regulation, apoptosis, and proliferation and is activated by high pressures in the vasculature (Lemarie et al., 2003), mechanical stretch in myocytes (Kumar and Boriek, 2003), and low amplitude cyclic strain in osteoblast‐like MF‐63 cells (Liu et al., 2007). Furthermore, direct links between PKC and NF‐kB activation have been documented in several cell lines (Sun and Yang, 2010). We hypothesized a link between extracellular pressure, calcium, and tumor proliferation.
We demonstrated that increased extracellular pressure‐stimulated proliferation in 3 colon cancer, a breast cancer, and 2 prostate cancer cell lines. The SW620 colon cancer cell line was chosen as a typical model for further study, and the studies were repeated after treatment with calcium chelators and calcium‐channel blockers. We identified a novel pressure‐sensitive calcium channel, Cav3.3, that drives proliferation by increasing [Ca2+]i. This Cav3.3‐dependent Ca2+ influx promotes proliferation through PKC‐β activation (not PKC‐α as previously suspected), which in turn mobilizes NF‐kB through the classical IKK‐IkB pathway. Pressure‐induced activation of these elements was Cav3.3‐dependent and ultimately increased cyclin D and proliferation. To assess the clinical relevance of our findings, we compared the lower pressure peripheries to the relatively higher pressure centers of 28 large primary human tumors and demonstrated gradients in IkB phosphorylation, NF‐kB, cyclin D, and proliferation in vivo consistent with the pathway delineated by our in vitro studies.
2. Materials and methods
Cells: Rat MAT‐Ly‐Lu‐B‐2 (MLL, ATCC CRL‐2376), murine CT‐26.CL25 (ATCC CRL‐2639) and human SW620 (ATCC CCL‐227), Caco‐2 C2BBe1 (ATCC CRL‐2102), MCF‐7 (ATCC HTB‐22) and PC‐3 (ATCC CRL‐1435) cancer cells were cultured by American Type Culture Collection (ATCC, Rockville, MD) recommendations. Cells were studied on collagen I‐precoated plates.
Pressure regulation: Pressure was manipulated for 24 h utilizing an airtight box with inlet and outlet valves for gas and manometry, as previously (Downey et al., 2008).
Proliferation: MTT absorbance was assayed per ATCC protocol. Briefly, we exposed 5000 cells/well in 96 well plates to increased or ambient pressure, added MTT reagents, and quantitated 570 nm absorbance. Control and pressure‐treated cells were also manually counted in 20 random fields of 6 well plates (Downey et al., 2008) with similar results.
Inhibitors: Extracellular and intracellular Ca2+ were chelated with 1 mM EGTA and 5 μM BAPTA‐AM, respectively (EMD Chemicals, Gibbstown, NJ). 10 μM lanthanum chloride (EMD Chemicals) blocked non‐specific divalent cation channels and 5 μM SKF96365 (Tocris Bioscience, Bristol, UK) blocked receptor‐mediated calcium‐entry (Merritt et al., 1990). Gadolinium chloride (Sigma–Aldrich, St. Louis, MO) inhibited stretch‐activated ion channels (Yang and Sachs, 1989). 5 μM nimodipine [77] blocked L‐type Ca2+ channels and 1 μM NNC 55‐0396 blocked T‐type channels (Tocris) (Huang et al., 2004). NiCl2 (Sigma–Aldrich) blocked T‐type channel subtypes, with 20 μM blocking Cav3.2 and 200 μM blocking Cav3.1 and Cav3.3 (Lee et al., 1999). 100 nM calphostin‐C blocked PKC globally, 6 nM GO6976 blocked PKC‐α and PKC‐β, 15 nM 3‐(1‐(3‐Imidazol‐1‐yl propyl)‐1H‐indol‐3‐yl)‐4‐anilino‐1H‐pyrrole‐2,5‐dione blocked PKC‐β alone, and 10 nM PKC‐ε translocation inhibitor peptide (EMD chemicals) blocked PKC‐ε. 10 mM IKK‐2 inhibitor [5‐(p‐fluorophenyl)‐2‐ureido]‐thiophene‐3‐carboxamide, 40 nM IKK‐3 inhibitor [5‐(5,6‐dimethoxybenzinidazol‐1‐yl)‐3‐(2‐methanesulfonyl‐benzyloxy)‐thiophene‐2‐carbonitrile], and a 90 nM IKK inhibitor N(6‐chloro‐9H‐β‐carbolin‐8‐yl)‐nicotinamide, that blocks IkB phosphorylation, were used per manufacturer's protocol separately and in combination (EMD Chemicals). 30 μM NSC23766 inhibited rac1 (EMD Chemicals). 65 nM PP2 (EMD Chemicals) was used for Src family inhibition, and Akt was inhibited using 1 μM Akt inhibitor IV (EMD Chemicals). 12 μM SN50 (EMD Chemicals) blocked NF‐kB p50 nuclear localization. An SN50 inactive analog that does not affect NF‐kB nuclear translocation was used as a control. 25 μM NF‐kB Serine 276 inhibitory peptide (Imgenex, San Diego, CA) acts as a p65 decoy through phosphorylation at that site. Cells were treated with the inhibitory peptide or an inactive control. 1 μM TCH 021, a novel imidazoline, inhibits NF‐kB gene transcription by modulating IkB degradation and subsequently inhibiting DNA binding (Kahlon et al., 2009; Peddibhotla and Tepe, 2004; Sharma et al., 2004). All inhibitors were diluted in sterile PBS, DMSO, or water and used for 24 h unless stated.
Small interfering RNA: Cav3.1, Cav3.3, PKC‐β, PKC‐α, and NF‐kB proteins were reduced using at least siRNA specific to each protein with similar results (Cell Signaling, Beverly, MA), oligofectamine, and Plus reagent (Invitrogen, Carlsbad, CA) by manufacturer's protocol, using non‐targeting controls in parallel (Dharmacon, Lafayette, CO). These experiments were performed 48 h after transfection.
Intracellular calcium visualization: Cells cultured at 70% confluence on glass coverslips (CS‐22/40, Warner Instruments, Hamden, CT) for 24 h were either treated with siRNA 24hr before plating or with PKC‐β inhibitor 30 min before visualization. Coverslips were then incubated in the dark for 30 min with fluorescent calcium‐sensitive dye, X‐rhod I AM (X‐1420, Invitrogen), prepared in Ringer's lactate, placed into the RC‐30 chamber (Warner Instruments), and subjected to ambient or 15 mmHg increased pressure. Pressure was manipulated by raising the reservoir of Ringer's lactate connected to the RC‐30 chamber to either the level of the chamber or 21 cm above it. Cells were visualized by fluorescent confocal microscopy.
Immunoprecipitation and western blotting: Cells were lysed, and protein was quantitated and resolved by SDS‐PAGE before western blotting on nitrocellulose (GE Healthcare, Little Chalfont, Buckinghamshire, UK) as previously (Downey et al., 2008). Coimmunoprecipitation studies were performed using 400 μg of protein with appropriate antibody and agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) as described (Downey et al., 2008). Rabbit polyclonal antibodies to pIKKα/β (ser180/181), NF‐kB p50 (Cell Signaling), total IkB (Sigma–Aldrich), Histone H1 (Santa Cruz) and NF‐kB p65 (Millipore, Billerica, MA), and appropriate anti‐rabbit secondary antibodies were used. Mouse monoclonal antibodies to cyclin D1, pIkB (ser 32/36, Cell Signaling), and actin (Sigma–Aldrich) were also used, with a horseradish peroxidase conjugated anti‐mouse secondary antibody. Protein was visualized by ECL‐Plus (GE Healthcare) and quantitated by Kodak Phosphorimager (Perkin Elmer, Boston, MA) within the linear range of exposure.
Nuclear fractionation: Nuclear fractions were obtained using the Qproteome nuclear subfractionation kit (Qiagen, Frederick, MD).
NF‐kB p50 and p65 transcription factor activity: We evaluated NF‐kB p50 and p65 DNA binding activity in nuclear lysates added to multi‐well plates coated with the double stranded DNA consensus sequence by an ELISA based NF‐kB p50 or p65 Transcription Factor Assay Kit (Cayman Chemical, Ann Arbor, MI).
NF‐kB activation: Cellular NF‐kB activation was assayed using a luciferase‐based NF‐kB lentiviral reporter assay (Qiagen). 5000 cells/well were plated in 96 well plates for 24 h and lentiviral particles were introduced with SureENTRY transduction reagent per manufacturer's protocol (Qiagen) for 24 h. The lentiviral suspension was replaced with normal medium, and cells were exposed to ambient or increased pressure for 24 h. Luciferin was added using the bright‐glow luciferase assay (Promega, Madison, WI) and luminescence quantitated by a FLUOstar Omega plate reader (BMG LabTech, Offenburg, Germany).
Flow cytometry: S‐phase fraction was measured in previously serum‐starved cells exposed to ambient or increased pressure for 24 h as previously (Walsh et al., 2004).
TUNEL staining: Apoptosis was evaluated by TUNEL staining (Roche Applied Science, Indianapolis, IN) per manufacturer's protocol. Apoptotic cells were counted on each slide, and control and pressure‐treated cells compared.
Active NF‐kB, cyclin D1 and IkB immunohistochemistry in human tumors: Under IRB‐approved protocol, archived colon, lung and head and neck malignant tumors were sectioned, deparaffinized, steamed at 95 °C with citrate antigen retrieval buffer (DAKO, Carpinteria, CA), rinsed with PBS and fixed with 3% hydrogen peroxide. Non‐specific staining was prevented by adding horse serum (Vector Laboratories, Burlingame, CA). The slides were rinsed and primary antibody to active NF‐kB (Invitrogen), cyclin D1, or IkB was added at room temperature. After PBS‐washing, slides were incubated with the biotinylated secondary antibody, streptavidin‐peroxidase and amino‐ethyl carbazol chromogen (VectaStain Universal Rapid Kit, Vector). Staining intensity was monitored to prevent overstaining. Slides were hematoxylin‐counterstained and coverslipped using Geltol (ThermoShandon, Fisher Scientific, Hanover Park, IL). An observer blinded to the study assigned scores from 0 (no immunostaining) to 4 (maximal immunostaining intensity) to the three areas under review; tumor center, tumor periphery and adjacent non‐malignant tissue. Areas were determined based on proximity to non‐malignant tissue, cell morphology and density. The entire area of each slide was assessed save for sections that were unsuitable for evaluation because of poor histological quality. Discernible mitotic figures were also counted in each area of the tumor periphery and center. Mitotic figures were also counted separately in tumor areas defined subjectively by a blinded reviewer as highly immunoreactive for active NF‐kB or less immunoreactive for active NF‐kB. All areas of the slide were counted for these studies.
Statistical Analysis: Differences between two groups were analyzed using Student's t‐test, and differences in immunostaining intensities by chi‐squared test. Statistical significance was set at p < 0.05.
3. Results
3.1. Increased extracellular pressure stimulates proliferation in colon, breast, and prostate cancer cells in vitro
SW620, Caco‐2, and CT‐26 colon; MCF‐7 breast; and MLL and PC3 prostate cancer cells were exposed to 0–80 mmHg increased extracellular pressure for 24 h and proliferative activity was assessed by MTT assay. Each cell line displayed increased MTT fluorescent intensity across the range of pressures, with greatest responses at 40–60 mmHg increased pressure (n = 6; p < 0.05, Figure 1). Parallel manual cell counting confirmed increased cell numbers after exposure to 40 mmHg increased pressure vs. ambient (n = 4; p < 0.05, not shown). Subsequent studies used even lower pressures to conservatively stay within the pathophysiologically relevant range.
Figure 1.
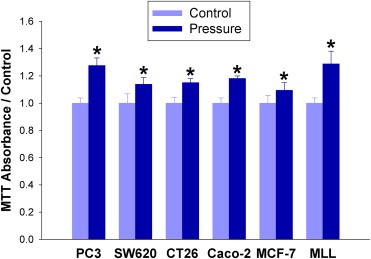
Extracellular pressure stimulates proliferation. PC3 prostate cancer, SW620, CT‐26, and Caco‐2 colon cancer; MCF‐7 breast cancer; and MLL prostate cancer cells were incubated at either ambient pressure or 40 mmHg increased pressure for 24 h and quantified by MTT assay. (*p < 0.05 vs. paired controls).
3.2. Ca2+ influx required for pressure‐stimulated proliferation in colon adenocarcinoma
The extracellular calcium chelator EGTA (1 mM) and intracellular chelator BAPTA‐AM (5 μM) each abolished the mitogenic effect of pressure, demonstrating the necessity of calcium for the effect (n = 8; p < 0.05 Figure 2A). The non‐specific divalent cation channel blocker lanthanum chloride (10 μM) and SKF96365 (5 μM), a blocker of receptor‐mediated calcium‐entry, differentiated between intracellular calcium increases due to receptor‐mediated and/or voltage‐gated channels from those due to release from internal stores. Like the chelators, each inhibitor of Ca2+ entry inhibited pressure‐driven proliferation (n = 8; p < 0.05 Figure 2A). Observing real‐time calcium flow under confocal microscopy with the calcium‐sensitive dye, X‐rhod‐1, we observed a distinct increase in [Ca2+]i following a 15 mmHg increase in hydrostatic pressure (n = 8; p < 0.05 Figure 2B).
Figure 2.
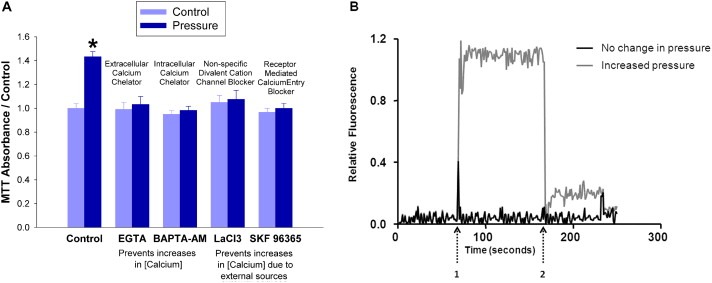
Extracellular pressure induces an influx of calcium that is required for pressure‐stimulated proliferation. (2A) SW620 cells were treated with either an extracellular calcium chelator (EGTA 1 mM), an intracellular chelator (BAPTA‐AM 5 μM), a non‐specific divalent cation channel blocker (lanthanum chloride 10 μM), or a blocker of receptor‐mediated calcium‐entry (SKF96365 5 μM) during 24 h exposure to ambient (light blue bars) or 15 mmHg increased extracellular pressure (dark blue bars). Cells quantified by MTT assay. (*p < 0.05 vs. paired controls) (2B) X‐rhod‐1, a fluorescent calcium‐sensitive dye, was added to SW620 cells 30 min before visualization by fluorescent confocal microscopy. The study began at ambient pressure which was increased transiently by 15 mmHg at arrow 1 and returned to baseline ambient pressure at arrow 2.
3.3. Pressure‐induced Ca2+ influx and proliferation dependent on T‐type Ca2+ channel Cav3.3
The proliferation assay was repeated in the presence of channel‐specific inhibitors. Gadolinium chloride, which inhibits stretch‐activated calcium channels, did not block the pressure effect at 100 μM, a concentration ten times higher than the reported IC50 (Yang and Sachs, 1989) (n = 8; p < 0.05, not shown). 5 μM nimodipine (IC50 = 3 μM), an L‐type Ca2+ blocker, was also unable to block the mitogenic effect of pressure, whereas 1 μM NNC 55‐0396 (IC50 = 7 μM), the T‐type channel blocker, negated the stimulation of proliferation by pressure (n = 8; p < 0.05 Figure 3A). Nickel chloride only blocked proliferation at concentrations of 100 μM or higher, suggesting that Cav3.1 or Cav3.3 might be the channel responsible, rather than Cav3.2 which is inhibited at 20 μM (Lee et al., 1999) (n = 8; p < 0.05 Figure 3B). Ca2+ visualization with confocal microscopy after treatment with channel‐specific siRNA revealed that the pressure‐driven influx of calcium persisted after ≈50% Cav3.1 reduction but was abolished by ≈50% Cav3.3 reduction (n = 8; p < 0.05, Figure 3C and D). Pressure‐induced proliferation despite ≈50% siRNA reduction of Cav3.1, or Cav3.2 but not after ≈50% Cav3.3 reduction (n = 12; p < 0.05, Figure 3E).
Figure 3.
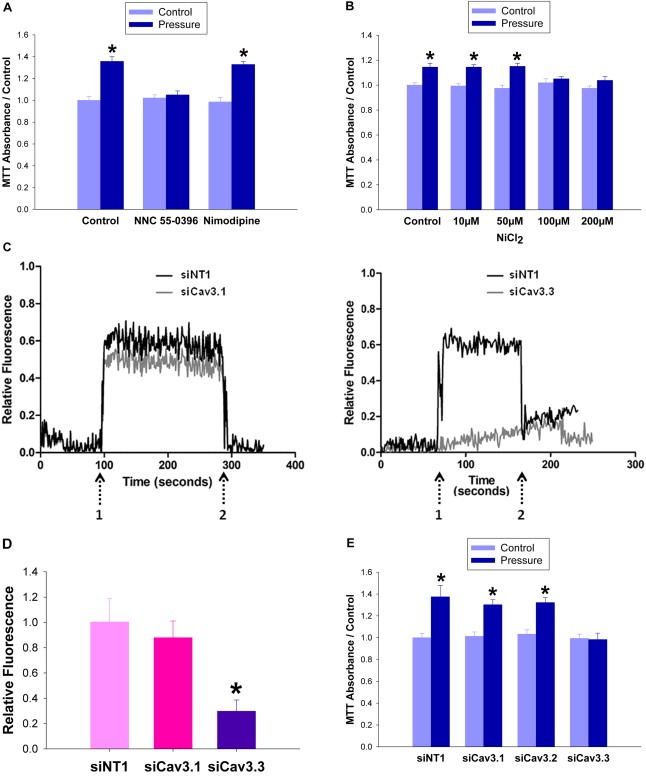
T‐type Ca 2+ channel Cav3.3 is necessary for pressure‐induced Ca 2+ influx and proliferation. (3A) SW620 cells treated with T‐type channel blocker (NNC 55‐0396 5 μM) or L‐type Ca2+ blocker (nimodipine 1 μM) were subjected to ambient pressure (light blue bars) or 15 mmHg increased extracellular pressure (dark blue bars) and MTT assay was performed at 24 h (3B) SW620 cells were treated with varying concentrations of nickel chloride to inhibit either Cav3.2 (IC50 20 μM) or Cav3.1 and 3.3 (IC50 200 μM) prior to pressurization to 15 mmHg. Both experiments show a loss of pressure‐induced proliferation with blockade of T‐type calcium channels Cav3.1 and 3.3. (3C) Cav3.1 or Cav3.3 was reduced in SW620 cells via siRNA. 48 h after transfection, Ca2+ was visualized within the cells by fluorescent confocal microscopy. The study began at ambient pressure which was increased transiently by 15 mmHg at arrow 1 and returned to baseline ambient pressure at arrow 2. (3D) The bar graph shows the area under the curve of the cumulative fluorescence seen in the Cav3.1 and 3.3 siRNA transfected cells vs. non‐targeting controls. (3E) SW620 cells were treated with siRNA specific to Cav3.1, 3.2, or 3.3, or with a non‐targeting control for 48 h before 24 h of incubation under ambient (light blue bars) or 15 mmHg increased pressure (dark blue bars). Cells were then quantified using an MTT reagent. (*p < 0.05 vs. paired controls).
3.4. Pressure‐induced Ca2+ influx activates PKC‐β to stimulate proliferation
The reliance of the pressure signal on [Ca2+]i increases led us to compare the effects of inhibiting Ca2+ dependent PKC‐α and PKC‐β with that of Ca2+‐independent PKC‐ε (Braz et al., 2002). Inhibiting either PKC‐α/β together or PKC‐β alone abolished the effect of pressure on proliferation (n = 9; p < 0.05, Figure 4A). Conversely, inhibition of Ca2+ independent PKC‐ε failed to do so. Similarly, siRNA knockdown of PKC‐β, but not PKC‐α, abolished pressure‐stimulated proliferation (n = 7; p < 0.05, Figure 4B) (n = 12; p < 0.05, Figure 4C). 15 mmHg pressure increased PKC‐β levels in the membrane fraction, and this was blocked by siRNA to Cav3.3 (n = 6; p < 0.05, Figure 4D). Alternatively, PKC‐β inhibition failed to attenuate the pressure‐induced calcium, consistent with PKC‐β being downstream of the Cav3.3‐induced calcium flux in the pressure pathway (n = 6; p < 0.05, Figure 4E).
Figure 4.
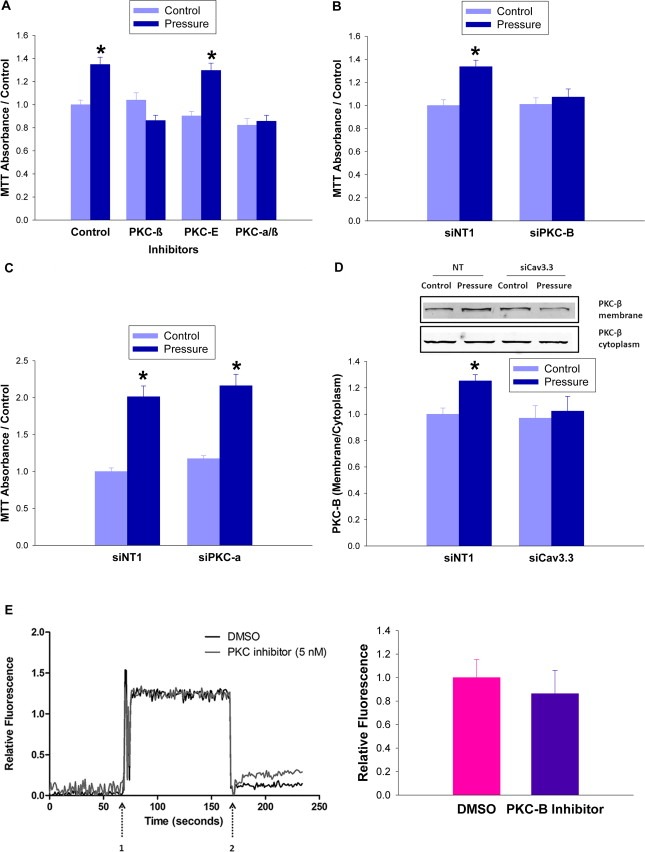
PKC‐β is activated by extracellular pressure. (4A) SW620 cells were treated with a PKC‐α/PKC‐β inhibitor (GO6976 6 nM), a PKC‐β specific inhibitor (3‐(1‐(3‐Imidazol‐1‐ylpropyl)‐1H‐indol‐3‐yl)‐4‐a nilino‐1H‐pyrrole‐2,5‐dione 15 nM), or a PKC‐ε specific inhibitor (PKC‐ε translocation inhibitor peptide10 nM) immediately before incubation at ambient (light blue bars) or 40 mmHg increased (dark blue bars) pressure for 24 h before MTT assay. (4B) Knockdown of PKC‐β was achieved by 48 h of siRNA transfection before 24 h of pressurization at ambient or 15 mmHg increased pressure and MTT assay. (4C) Knockdown of PKC‐α was achieved by 48 h of siRNA transfection before 24 h of pressurization at ambient or 15 mmHg increased pressure and MTT assay. (4D) SW620 cells were transfected for 48 h with siRNA against Cav3.3 or with a non‐targeting control before being exposed to 15 mmHg pressure. PKC‐β levels in the membrane fraction were measured after 24 h (4E) SW620 cells treated with PKC‐β inhibitor (5 nM) immediately prior to Ca2+ visualization by fluorescent confocal microscopy. The study began at ambient pressure which was increased transiently by 15 mmHg at arrow 1 and returned to baseline ambient pressure at arrow 2. The bar graph shows the area under the curve of the cumulative fluorescence in the DMSO and PKC‐β inhibitor treated groups. (*p < 0.05 vs. paired controls).
3.5. Cav3.3 required for pressure‐activation cascade of IKK, IkB, and NF‐kB
NF‐kB is sequestered within the cytosol by the binding of its inhibitor IkB. When IkB is phosphorylated by the kinase IKK, it releases NF‐kB, exposing the NF‐kB nuclear localization signal (Lee et al., 2007; Yamamoto and Gaynor, 2001). IKK phosphorylation was increased 27 ± 4% in pressure‐treated cells relative to ambient pressure control cells (n = 8; p < 0.05, not shown). IkB phosphorylation also increased 61 ± 8% in pressure‐treated cells (n = 8; p < 0.05, Figure 5A), but not after siRNA reduction of Cav3.3 (n = 7; p < 0.05, Figure 5B). Pressure decreased NF‐kB‐IkB association 31 ± 4% (n = 5; p < 0.05, Figure 5C). NF‐kB translocation was confirmed via measuring p50 and p65 subunit levels in nuclear fractions, which increased 58 ± 6% and 67 ± 8% respectively in pressure‐treated versus ambient pressure cells (n = 6; p < 0.05, Figure 5D). ELISA based measurements of NF‐kB p50 and p65 transcription factor activity showed increases of 50 ± 4% and 48 ± 6% respectively in response to pressure that were reversed with siCav3.3 treatment (n = 6; p < 0.05, Figure 5E). To assess the effect of IKK on pressure‐stimulated NF‐kB activation we treated SW620 cells with NF‐kB lentiviral reporter particles expressing firefly luciferase. We incubated them under ambient or 40 mmHg increased pressure for 24 h in the presence of inhibitors to IKK‐2 or IKK‐3, or an IKK inhibitor that blocks IkB phosphorylation. Individually, these inhibitors did not prevent pressure activation of NF‐kB (n = 12; p < 0.05, not shown), but the combination of the IKK‐2 and IKK‐3, the IKK‐2 and global IKK, or the three inhibitors used together each abolished pressure activation of NF‐kB (n = 12; p < 0.05, Figure 5F).
Figure 5.
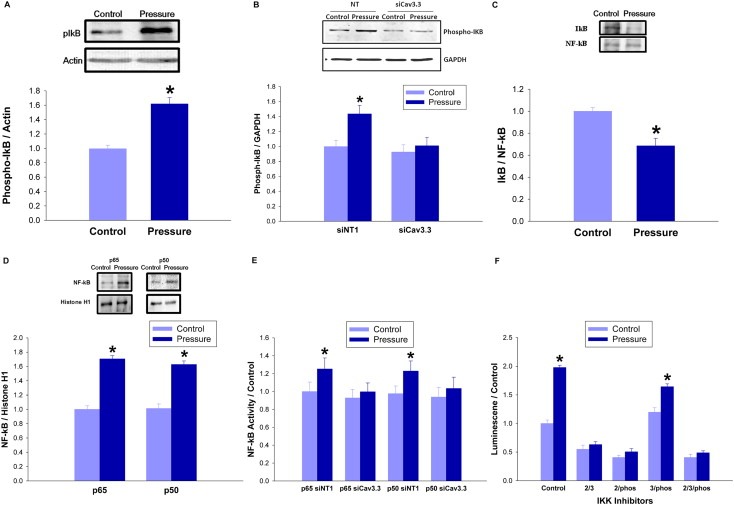
Pressure activates the IKK–IkB–NF‐kB signaling cascade in a Cav3.3‐dependent manner. (5A) SW620 cells were exposed to 40 mmHg for 24 h before Western blotting with phospho‐IkB antibodies. Densitometric results were normalized to actin. (5B) SW620 cells were transfected with siRNA targeting Cav3.3 or with a non‐targeting control siRNA for 48 h and then incubated under ambient or 15 mmHg increased pressure for 24 h. Western blots with phospho‐IkB antibodies were analyzed and normalized to GAPDH. (5C) Lysate from SW620 cells incubated at ambient or 40 mmHg increased pressure for 24 h was immunoprecipitated with antiNF‐kB antibodies and the resulting immunoprecipitates were immunoblotted with IkB antibodies to identify IkB associated with NF‐kB. (5D) Nuclear fractions from SW620 cells that had been incubated at ambient or 40 mmHg increased pressure for 24 h were immunoblotted with antibodies against the p65 and p50 subunits of NF‐kB. Histone H1 served as a loading control. (5E) Lysate from SW620 cells transfected with siRNA targeting Cav3.3 or with a non‐targeting control for 48 h and then incubated at ambient or 15 mmHg increased pressure for 24 h was then used to quantify NF‐kB p65 and p50 subunit activity by ELISA. (5F) SW620 cells were treated with NF‐kB lentiviral reporter particles expressing firefly luciferase and incubated under ambient or 40 mmHg increased pressure for 24 h in the presence of an IKK‐2 inhibitor ([5‐(p‐fluorophenyl)‐2‐ureido]‐thiophene‐3‐carboxamide, 10 mM), an IKK‐3 inhibitor ([5‐(5,6‐dimethoxybenzinidazol‐1‐yl)‐3‐(2‐methanesulfonyl‐benzyloxy)‐thiophene‐2‐carbonitrile] 40 nM), or an IKK inhibitor that blocks IkB phosphorylation (N(6‐chloro‐9H‐β‐carbolin‐8‐yl)‐nicotinamide 90 nM). (*p < 0.05 vs. paired controls).
3.6. Pressure activates NF‐kB through PKC‐β
Pressure increased NF‐kB activation 94 ± 7% (n = 12; p < 0.05, Figure 6A). The NF‐kB nuclear decoy and the NF‐kB inhibitors SN50 and TCH 021 each prevented pressure‐associated NF‐kB activation (n = 6; p < 0.05, Figure 6A). FAK and Akt facilitate the effects of extracellular pressure on integrin‐mediated adhesion in colon cancer cells (Basson, 2008), and FAK also stimulates proliferation in response to another physical force, cyclic deformation (Li et al., 2001). However, neither FAK nor Akt inhibition prevented pressure activation of NF‐kB (n = 9; p < 0.05, Figure 6B). Conversely, the global PKC inhibitor calphostin‐C, the PKC‐α/β inhibitor Go6976, and a pure PKC‐β inhibitor all abolished the effect of pressure on NF‐kB activation, while PKC‐ε inhibition did not (n = 9; p < 0.05, Figure 6C). Consistent with the chemical inhibitor data, siRNA knockdown of PKC‐β reversed the effect (n = 6; p < 0.05, Figure 6D). The pressure‐driven increase in NF‐kB activation was also abolished by siRNA to Cav3.3 (n = 7; p < 0.05, Figure 6D).
Figure 6.
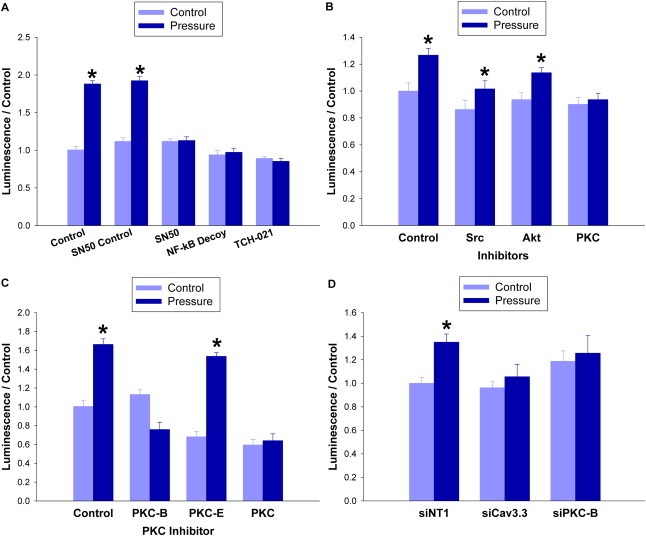
Pressure activates NF‐kB in a PKC‐β dependent manner. (6A) NF‐kB activation was measured by treating SW620 cells with NF‐kB lentiviral reporter particles expressing firefly luciferase. They were then incubated under ambient or 40 mmHg increased pressure for 24 h in the presence of NF‐kB inhibitor SN50 (12 μM), or its control peptide, NF‐kB nuclear decoy (25 μM), or NF‐kB inhibitor TCH 021 (1 μM). (6B) NF‐kB activation was also measured in cells treated with an inhibitor to Src (PP2 65 nM), Akt (Akt inhibitor IV 1 μM), or PKC (calphostin 100 nM) before undergoing 24 h of ambient or 40 mmHg pressure exposure. (6C) The experiment was repeated using the global PKC inhibitor as well as inhibitors specific for PKC‐β (3‐(1‐(3‐Imidazol‐1‐yl propyl)‐1H‐indol‐3‐yl)‐4‐anilino‐1H‐pyrrole‐2,5‐dione 15 nM) and PKC‐ε (PKC‐ε translocation inhibitor peptide 10 nM). (6D) SW620 cells were transfected for 48 h with siRNA against Cav3.3, PKC‐β, or with a non‐targeting control before being exposed to ambient or 15 mmHg pressure. NF‐kB activation was measured via lentiviral reporter particles expressing firefly luciferase. (*p < 0.05 vs. paired controls).
3.7. Pressure stimulates proliferation through NF‐kB
To connect NF‐kB to the pressure proliferation effect, we attempted to block the increase using NF‐kB inhibitors as well as an inhibitor of another mitogen signal, rac1 for comparison. The rac1 inhibitor NSC23766 did not prevent increased MTT fluorescent intensity in pressure‐treated cells, but the NF‐kB inhibitor SN50, an NF‐kB nuclear decoy, and a novel imidazoline inhibitor, TCH 021, abolished the effect (n = 4; p < 0.05, Figure 7A). Flow cytometry showed S‐phase fraction increases of 22 ± 6% in cells exposed to increased pressure (n = 3; p < 0.01, Figure 7B) but not in those treated with inhibitors as well (n = 4; p < 0.05, Figure 7B). TUNEL staining demonstrated that pressure did not alter cell apoptosis (n = 3; p < 0.05, not shown). A measurable product of the mitogenic effect of NF‐kB is the accumulation of the cell‐cycle regulator Cyclin D1, which increased in pressure‐treated cells that did not undergo siRNA Cav3.3 treatment (n = 6; p < 0.05, Figure 7C).
Figure 7.
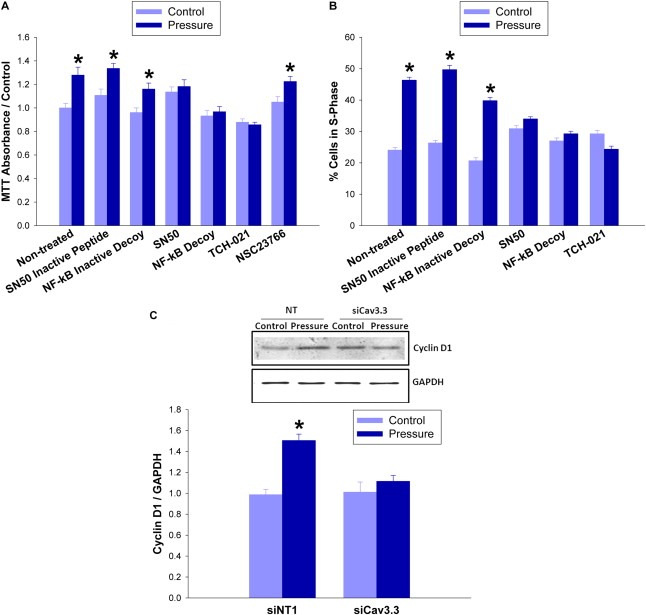
Pressure induces proliferation through Cav3.3 and NF‐kB. (7A) SW620 cells were incubated under ambient or 40 mmHg increased pressure for 24 h in the presence of an NF‐kB inhibitor SN50 (12 μM) or its control peptide, an NF‐kB nuclear decoy (25 μM) or its inactive control, an NF‐kB inhibitor TCH 021 (1 μM), or a rac1 inhibitor (NSC23766 30 μM) before MTT assay. (7B) S‐phase fraction was measured in previously serum‐starved SW620 cells via flow cytometry. Before measurement, cells were incubated under ambient or 40 mmHg increased pressure for 24 h in the presence of an NF‐kB inhibitor SN50 (12 μM) or its control peptide, an NF‐kB nuclear decoy (25 μM) or its inactive control, an NF‐kB inhibitor TCH 021 (1 μM). (7C) SW620 cells were transfected with siRNA targeting Cav3.3 or with a non‐targeting control for 48hr and exposed to ambient or 15 mmHg for 24hr before being lysed and immunoblotted with cyclin D1 antibodies and normalized to GAPDH. (*p < 0.05 vs. paired controls).
3.8. NF‐kB, IkB and cyclin D1 staining increases in the center of human tumors
Since extracellular interstitial pressures are higher in the center of large solid human tumors than at their peripheries (Boucher et al., 1990; Sarntinoranont et al., 2003), we compared mitotic rates at the periphery and center of 28 human colon, lung, and head and neck tumors, averaging 8 ± 1.2 cm diameter. A reviewer blinded to the hypothesis reported a 154 ± 14% increase in mitotic figures per hpf in the central regions of the tumors relative to their peripheries (n = 15; p < 0.05, Figure 8D). Parallel studies demonstrated high immunoreactivity for active NF‐kB, phospho‐IkB, and cyclin D1 in the centers of these tumors which decreased towards their peripheries. Little or no staining was observed in adjacent non‐malignant tissues from each specimen. (n = 27; p < 0.05, Figure 8A–C). Assessment of immunoreactivity on a scale from 0 (negative staining) to 4 (high staining), specimens immunostained for NF‐kB were scored in 95 zones from adjacent histologically normal margins from 19 different tumor samples and averaged a score of 0.8. Blinded evaluation of areas at the periphery of the tumors in these same slides yielded a mean score of 2.2, while central tumor zones received a mean score of 3.6. Similar gradients occurred for phospho‐IkB and cyclin D1 immunoreactivity (Table 1). Differences between the normal tissue, the peripheral zone, and the central tumor zone were each statistically significant (p < 0.01). Mitotic figures were separately counted in 20 fields of highly NF‐kB‐immunoreactive areas within the tumors compared to less NF‐kB‐immunoreactive areas, confirming a 124 ± 8% increase in mitotic figures within these more immunoreactive zones (p < 0.05, not shown).
Figure 8.
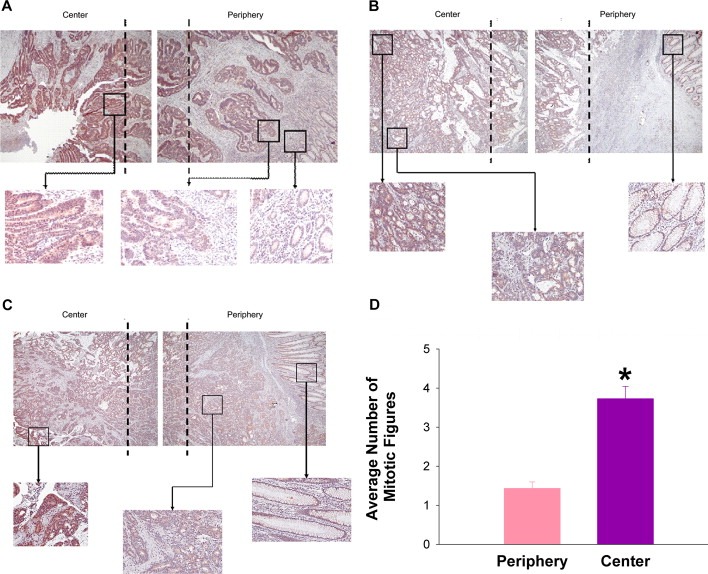
Immunohistochemical staining of colorectal carcinomas. Staining of colorectal carcinomas was representative of that observed in colorectal, head and neck and lung tumors. To obtain a full view of the sample, a panel of overlapping photographs were taken and placed together. Dashed lines represent zones of overlap within the image. Overall panel images were taken at 40× magnification and higher power images within each zone at 200×. NF‐kB (8A), phospho‐IkB (8B), and cyclin D1 (8C) immunoreactivity and mitotic index (8D) are increased in tumor centers vs. peripheries. Non‐malignant tissue showed minimal staining and few mitotic figures.
Table 1.
Immunohistochemical scores.
| Antibody | Cases | Average intensity (number of zones counted) | p‐value | ||
|---|---|---|---|---|---|
| Non‐malignant | Periphery | Center | |||
| NF‐kB | 19 | 0.8 (95) | 2.2 (95) | 3.6 (95) | <0.01 |
| Phospho‐IkB | 23 | 0.6 (115) | 2.0 (115) | 3.4 (115) | <0.001 |
| Cyclin D1 | 21 | 0.9 (105) | 2.4 (105) | 3.6 (105) | <0.01 |
Immunohistochemical staining of colorectal carcinomas, representative of staining observed in colorectal, head and neck and lung tumors. Mean immunohistochemical NF‐kB, phospho‐IkB, and cyclin D1 intensity in tumor centers vs. peripheries vs. adjacent non‐malignant tissue, scored by a blinded observer.
4. Discussion
This study suggests that increased extracellular pressure stimulates tumor proliferation by activating the T‐type Ca2+ channel Cav3.3. The consequent calcium influx activates a PKC‐β‐dependent pathway, phosphorylation of IKK and IkB, NF‐kB activation, and proliferation. These results implicate a calcium channel that is not thought stretch‐activated as a primary mechanoreceptor, and suggest a specificity for PKC‐β. Finally, since we studied pressures similar to large human tumors (Boucher et al., 1990; Gutmann et al., 1992; Less et al., 1992; Raju et al., 2008) and since tumor periphery‐center gradients of proliferation and related signals parallel previous observations of similar gradients in interstitial pressures, our results suggest that in vivo such pressures stimulate cancer proliferation, causing tumor expansion that further increases pressure in a vicious positive feedback cycle that may potentiate cancer growth.
Existing models of pressure signaling split between two theories of mechanoperception. Intrinsic mechanosensitivity relies on tension along the lipid membrane to drive protein subunit recruitment or realignment to affect ion channel conductance (Hamill and McBride, 1994; Martinac et al., 1990; Opsahl and Webb, 1994). Extrinsic mechanosensitivity relays mechanical stress via cytoskeletal or extracellular elements to cell enzymes (Guharay and Sachs, 1984; Hamill and McBride, 1992; Ingber, 1997). Early mechanosensitive channels were characterized under negative pressures as intrinsic stretch‐activated channels that opened with lateral membrane tension but resisted the perpendicular force of hydrostatic pressure (Sokabe et al., 1991). The volumetrically incompressible lipid bilayer poorly conducts forces generated by tumor growth (Hamill and Martinac, 2001). Thus, mitogenic pressure signals might be postulated to require cytoskeletal sensing, consistent with pressure effects on migration and adhesion (Kovalenko et al., 2012; Thamilselvan and Basson, 2004). However, phalloidin, which inhibits actin depolymerization (Cooper, 1987), attenuates pressure effects on adhesion but not proliferation (Basson, 2008). While actin depolymerization occurs within minutes during adhesion (Wang et al., 1993) and is necessary for pressure‐stimulated adhesion by suspended cells (Thamilselvan and Basson, 2004), cytoskeletal rearrangement seems less likely to be required for pressure to stimulate proliferation in adherent cells.
Pressure requires 4–6 h to commit adherent cells to increased proliferation (Walsh et al., 2004) but stimulates adhesiveness within 1 min in suspended cells (Craig et al., 2009). The latter requires cytoskeletal mechanosensing, PI3K, Src, FAK, Akt and rac1 (Basson, 2008; Downey et al., 2008). However, the mitogenic effects of pressure are independent of Src, PI‐3‐kinase and actin depolymerization (Basson, 2008; Walsh et al., 2004) and we show here FAK, AKT and rac1 independence. This may reflect differences in the intracellular kinome of adherent cells. Internal pre‐stress remodels the cytoskeletons of suspended cells into spheres, altering the kinetics of attached enzymes (Ingber, 1997). Adherent cells, however, are isometrically balanced between inward pull by the contractile cytoskeleton and outward pull by the extracellular matrix and adjacent cells, and thus experience a different kinomic landscape (Ingber, 1997). Cells experiencing such tensegrity forces are more responsive to mitogens (Ingber, 2008) and sites of high mechanical stress yield higher growth rates in vitro (Nelson et al., 2005). Pressure may also be a mitogen better perceived by adherent cells. That the mitogenic effects of pressure in adherent cells require α‐actinin‐4 while the adhesiogenic effects of pressure in nonadherent cells require α‐actinin‐1(Craig et al., 2007a; Downey et al., 2011) may offer a clue to this difference in cytoarchitectural mechanotransduction.
While a single mechanical stimulus can effect different changes in a cell, different mechanical forces may conversely stimulate the same process by different pathways. Cyclic strain stimulates proliferation in human Caco‐2 colon cancer cells, H‐441 lung epithelial cells, and non‐malignant IEC‐6 enterocytes via a cytoskeleton‐dependent pathway involving PKC, Src, rac1, FAK and ERK (Chaturvedi et al., 2007, 2007, 1998, 2001), but none except PKC are required for the mitogenic effects of pressure (Kovalenko et al., 2012). Interestingly, cyclic strain stimulates Caco‐2 cancer cell proliferation in adherent cells via α‐actinin‐1(Craig et al., 2007b), so different forces also likely require different mechanotransduction.
Stretch‐activated ion channels also implicated in mechanotransduction and proliferation (Hamill and Martinac, 2001; Liu et al., 1994) activate under negative pressure and are inhibited by gadolinium, which stunts the lipid bilayer's ability to transmit lateral tension (Ermakov et al., 2001; Tanaka et al., 2002). However, gadolinium did not block pressure‐induced proliferation, echoing Cav3.3 stretch insensitivity (Calabrese et al., 2002). We believe this pressure‐activated Cav3.3‐PKC‐β‐NF‐kB pathway is novel among reported mechanisms for mechanically‐stimulated proliferation.
We show here that the non‐stretch activated T‐type calcium channel Cav3.3 responds to increased pressure and stimulates proliferation. Most mechanosensitive ion channels such as L‐type calcium channels (Kraichely and Farrugia, 2007) are intrinsically mechanosensitive and exhibit stretch‐activation under negative pressures, Some calcium permeable channels may respond to positive pressure in rat endocardial endothelium, but even these are stretch‐activated (Kohler et al., 1998). We found that the mitogenic effects of pressure required an extracellular calcium influx, and. despite the lack of reports of T‐type Ca2+ channel mechanoperception, Cav3.3 indeed mediated calcium influx in response to pressure. Cav3.3 is mostly in neuronal cells and not known to be overexpressed in tumors (Lu et al., 2008), while Cav3.1 and Cav3.2are overexpressed in breast cancer, retinoblastoma and glioma (Bertolesi et al., 2002). However, neither L‐type channels nor Cav3.1/Cav3.2 seem required for pressure‐induced proliferation. Although T‐type Cav channels have previously been studied in the context of neural function, these findings suggest Cav3.3 and calcium signaling also mediate tumor growth.
PKC influences colon cancer cells (Basson and Hong, 1995), and several PKC isoforms respond to mechanical forces, but most are calcium‐independent, and PKC‐β has not been implicated to date. The calcium‐independent isozymes PKC‐δ, ε, θ and η are activated by diacylglycerol alone, whereas the isozymes PKC‐α, βΙ, βΙΙ and γ also require calcium (Mochly‐Rosen et al., 2012). Cyclic strain induces activation and membrane translocation of PKC‐α and ‐ζ in Caco‐2 cells (Han et al., 1998), and endothelial cells where cyclic strain does not translocate PKC‐β to the membrane (Cheng et al., 2001). Shear stress activates endothelial ERK by PKC‐ε, but not PKC‐α or ‐ζ (Traub et al., 1997), and osteoblast IL‐11 expression is stimulated by PKC‐δ (Kido et al., 2010). Interestingly, pressure stimulates integrin‐mediated adhesion independently of PKC in suspended Caco‐2 and SW620 cells (Thamilselvan and Basson, 2004). However, the same pressure applied to adherent colon cancer cells stimulates both PKC‐dependent proliferation and PKC‐α membrane translocation (Walsh et al., 2004). Here, consistent with the calcium‐dependent mitogenic effect of pressure, specific inhibition of calcium‐dependent PKC‐β prevented the mitogenic effects of pressure.
Pressure PKC‐β‐dependently activated IkB, IKK, and NF‐kB, consistent with previous observations that PKC‐β mediates NF‐kB activation (Sommer et al., 2005), although the opposite effect has been reported in human umbilical vein endothelial cells. Both hyperphosphatemia and hypophosphatemia increase PKCβII but decrease activated NF‐kB (Peng et al., 2011). Traditionally, IKK and IkB phosphorylation release NF‐kB to translocate the nucleus where it is mitogenic (Dolcet et al., 2005). However, NF‐kB can also kill cells by interacting with anti‐apoptotic factors like FLIP and TRAF1/2 (Dolcet et al., 2005), and causes colon cancer xenograft apoptosis (Stark et al., 2007). IKK‐2 ablation similarly causes myocyte proliferation (Mourkioti et al., 2006) but IKK‐2 is required for TNF‐α‐induced proliferation in human mesenchymal stem cells (Bocker et al., 2008). Thus, it was not obvious that NF‐kB activation would stimulate cancer cell proliferation in response to pressure before these studies. That the inhibitors of IKK‐2, IKK‐3 and IKK phosphorylation of IkB could only prevent pressure activation of NF‐kB when used in combination suggests pathway redundancy and confirms IKK relevance.
Intratumoral pressure is generated by changes in tumor volume, localized vascular hyperpermeability, lymphatic obstruction, and changes in the composition of the surrounding stroma (Ariffin et al., 2014). While most investigations have focused on intratumoral pressure, some tumors may also experience intermittent deformation and strain engendered by changes in blood pressure or movement, so that the mitogenic effects of pressure could act synergistically with the mitogenic effects of cyclic strain. Extracellular pressure increases also stimulate macrophage phagocytosis and cytokine release by a very different pathway (Shiratsuch and Basson, 2005; Shiratsuchi and Basson, 2004) and such modulation of immune function by intratumoral pressures could also affect tumor biology. These interesting questions await further study.
We studied pressures resembling the 4–50 mmHg average internal pressures of large human tumors in vivo (Gutmann et al., 1992; Less et al., 1992; Raju et al., 2008). In vivo measurements delineated mean tumor interstitial fluid pressures of 18 mmHg and 21 mm Hg in cervical (Milosevic et al., 2014), and colorectal cancers (Less et al., 1992). We studied 40 mmHg to elicit maximal proliferation and 15 mmHg to simulate relevant pathophysiology. The significance of elevated pressure and NF‐kB activation was validated by the gradient of phospho‐NF‐kB, IkB, and cyclin D1 immunoreactivity from the high pressure center towards the lower pressure periphery of large human tumors until nearly disappearing in the adjacent normal tissue. Indeed, NF‐kB immunoreactivity of the magnitude observed correlates with poor prognosis (Ismail et al., 2004).
Pathophysiologically relevant pressure increases stimulate colon, breast and prostate cancer proliferation by a Cav3.3‐dependent mechanism involving activation of PKC‐β and the IKK complex, NF‐kB p50 and p65 nuclear localization and activation, and increased cyclin D1 expression. How Cav3.3 senses pressure awaits further study, but the pressure generated by tumor growth against a stiff surrounding stroma may stimulate tumor proliferation, eliciting an positive feedback loop that stimulates tumor growth with unfortunate clinical consequences. The subsequent pressure increases may also impede tumor perfusion by drugs (Ariffin et al., 2014). Although it may be possible to decrease intratumoral pressure directly (Ariffin et al., 2014), this Cav3.3‐dependent pathway may represent a target of opportunity to slow tumor growth in patients not candidates for resection or conventional cytotoxics and/or to decrease intratumoral pressure by slowing tumor growth to facilitate conventional anti‐tumor therapy.
Acknowledgments
The technical assistance of Drs. David Craig, James Hatfield, and William Jackson is gratefully acknowledged. This work was supported by the National Institutes of Health (RO1DK060771 to M.D.B.).
Basson Marc D., Zeng Bixi, Downey Christina, Sirivelu Madhu P., Tepe Jetze J., (2015), Increased extracellular pressure stimulates tumor proliferation by a mechanosensitive calcium channel and PKC‐β, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.10.008.
References
- Ariffin, A.B. , Forde, P.F. , Jahangeer, S. , Soden, D.M. , Hinchion, J. , 2014. Releasing pressure in tumors: what do we know so far and where do we go from here? A review. Cancer Res. 74, 2655–2662. [DOI] [PubMed] [Google Scholar]
- Basson, M.D. , 2008. An intracellular signal pathway that regulates cancer cell adhesion in response to extracellular forces. Cancer Res. 68, 2–4. [DOI] [PubMed] [Google Scholar]
- Basson, M.D. , Hong, F. , 1995. Modulation of human CACO-2 intestinal epithelial cell phenotype by protein kinase C inhibitors. Cell Biol. Int. 19, 1025–1032. [DOI] [PubMed] [Google Scholar]
- Bertolesi, G.E. , Shi, C. , Elbaum, L. , Jollimore, C. , Rozenberg, G. , Barnes, S. , Kelly, M.E. , 2002. The Ca(2+) channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol. Pharmacol. 62, 210–219. [DOI] [PubMed] [Google Scholar]
- Bevan, R.D. , 1976. An autoradiographic and pathological study of cellular proliferation in rabbit arteries correlated with an increase in arterial pressure. Blood Vessels. 13, 100–128. [DOI] [PubMed] [Google Scholar]
- Bocker, W. , Docheva, D. , Prall, W.C. , Egea, V. , Pappou, E. , Rossmann, O. , Popov, C. , Mutschler, W. , Ries, C. , Schieker, M. , 2008. IKK-2 is required for TNF-alpha-induced invasion and proliferation of human mesenchymal stem cells. J. Mol. Med. (Berl). 86, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Boucher, Y. , Baxter, L.T. , Jain, R.K. , 1990. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 50, 4478–4484. [PubMed] [Google Scholar]
- Braz, J.C. , Bueno, O.F. , De Windt, L.J. , Molkentin, J.D. , 2002. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2). J. Cell Biol. 156, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese, B. , Tabarean, I.V. , Juranka, P. , Morris, C.E. , 2002. Mechanosensitivity of N-type calcium channel currents. Biophys. J. 83, 2560–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capiod, T. , Shuba, Y. , Skryma, R. , Prevarskaya, N. , 2007. Calcium signalling and cancer cell growth. Subcell. Biochem. 45, 405–427. [DOI] [PubMed] [Google Scholar]
- Chaturvedi, L.S. , Marsh, H.M. , Basson, M.D. , 2007. Src and focal adhesion kinase mediate mechanical strain-induced proliferation and ERK1/2 phosphorylation in human H441 pulmonary epithelial cells. Am. J. Physiol. Cell Physiol. 292, C1701–C1713. [DOI] [PubMed] [Google Scholar]
- Chaturvedi, L.S. , Marsh, H.M. , Shang, X. , Zheng, Y. , Basson, M.D. , 2007. Repetitive deformation activates focal adhesion kinase and ERK mitogenic signals in human Caco-2 intestinal epithelial cells through Src and Rac1. J. Biol. Chem. 282, 14–28. [DOI] [PubMed] [Google Scholar]
- Cheng, J.J. , Wung, B.S. , Chao, Y.J. , Wang, D.L. , 2001. Sequential activation of protein kinase C (PKC)-alpha and PKC-epsilon contributes to sustained Raf/ERK1/2 activation in endothelial cells under mechanical strain. J. Biol. Chem. 276, 31368–31375. [DOI] [PubMed] [Google Scholar]
- Cooper, J.A. , 1987. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 105, 1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig, D.H. , Gayer, C.P. , Schaubert, K.L. , Wei, Y. , Li, J. , Laouar, Y. , Basson, M.D. , 2009. Increased extracellular pressure enhances cancer cell integrin-binding affinity through phosphorylation of beta1-integrin at threonine 788/789. Am. J. Physiol. Cell Physiol. 296, C193–C204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig, D.H. , Haimovich, B. , Basson, M.D. , 2007. Alpha-actinin-1 phosphorylation modulates pressure-induced colon cancer cell adhesion through regulation of focal adhesion kinase-Src interaction. Am. J. Physiol. Cell Physiol. 293, C1862–C1874. [DOI] [PubMed] [Google Scholar]
- Craig, D.H. , Zhang, J. , Basson, M.D. , 2007. Cytoskeletal signaling by way of alpha-actinin-1 mediates ERK1/2 activation by repetitive deformation in human Caco2 intestinal epithelial cells. Am. J. Surg. 194, 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolcet, X. , Llobet, D. , Pallares, J. , Matias-Guiu, X. , 2005. NF-kB in development and progression of human cancer. Virchows Arch. 446, 475–482. [DOI] [PubMed] [Google Scholar]
- Downey, C. , Craig, D.H. , Basson, M.D. , 2008. Pressure activates colon cancer cell adhesion via paxillin phosphorylation, Crk, Cas, and Rac1. Cell Mol. Life Sci. 65, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey, C. , Craig, D.H. , Basson, M.D. , 2011. Isoform-specific modulation of pressure-stimulated cancer cell proliferation and adhesion by alpha-actinin. Am. J. Surg. 202, 520–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermakov, Y.A. , Averbakh, A.Z. , Yusipovich, A.I. , Sukharev, S. , 2001. Dipole potentials indicate restructuring of the membrane interface induced by gadolinium and beryllium ions. Biophys. J. 80, 1851–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guharay, F. , Sachs, F. , 1984. Stretch-activated single ion channel currents in tissue-cultured embryonic chick skeletal muscle. J. Physiol. 352, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann, R. , Leunig, M. , Feyh, J. , Goetz, A.E. , Messmer, K. , Kastenbauer, E. , Jain, R.K. , 1992. Interstitial hypertension in head and neck tumors in patients: correlation with tumor size. Cancer Res. 52, 1993–1995. [PubMed] [Google Scholar]
- Hamill, O.P. , Martinac, B. , 2001. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 81, 685–740. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P. , McBride, D.W. , 1992. Rapid adaptation of single mechanosensitive channels in Xenopus oocytes. Proc. Natl. Acad. Sci. U S A. 89, 7462–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill, O.P. , McBride, D.W. , 1994. The cloning of a mechano-gated membrane ion channel. Trends Neurosci. 17, 439–443. [DOI] [PubMed] [Google Scholar]
- Han, O. , Li, G.D. , Sumpio, B.E. , Basson, M.D. , 1998. Strain induces Caco-2 intestinal epithelial proliferation and differentiation via PKC and tyrosine kinase signals. Am. J. Physiol. 275, G534–G541. [DOI] [PubMed] [Google Scholar]
- Huang, L. , Keyser, B.M. , Tagmose, T.M. , Hansen, J.B. , Taylor, J.T. , Zhuang, H. , Zhang, M. , Ragsdale, D.S. , Li, M. , 2004. NNC 55-0396 [(1S,2S)-2-(2-(N-[(3-benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluoro-1,2, 3,4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride]: a new selective inhibitor of T-type calcium channels. J. Pharmacol. Exp. Ther. 309, 193–199. [DOI] [PubMed] [Google Scholar]
- Ingber, D.E. , 1997. Tensegrity: the architectural basis of cellular mechanotransduction. Annu. Rev. Physiol. 59, 575–599. [DOI] [PubMed] [Google Scholar]
- Ingber, D.E. , 2008. Can cancer be reversed by engineering the tumor microenvironment?. Semin. Cancer Biol. 18, 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail, H.A. , Lessard, L. , Mes-Masson, A.M. , Saad, F. , 2004. Expression of NF-kappaB in prostate cancer lymph node metastases. Prostate. 58, 308–313. [DOI] [PubMed] [Google Scholar]
- Kahlon, D.K. , Lansdell, T.A. , Fisk, J.S. , Tepe, J.J. , 2009. Structural-activity relationship study of highly-functionalized imidazolines as potent inhibitors of nuclear transcription factor-kappaB mediated IL-6 production. Bioorg. Med. Chem. 17, 3093–3103. [DOI] [PubMed] [Google Scholar]
- Kawata, Y. , Mizukami, Y. , Fujii, Z. , Sakumura, T. , Yoshida, K. , Matsuzaki, M. , 1998. Applied pressure enhances cell proliferation through mitogen-activated protein kinase activation in mesangial cells. J. Biol. Chem. 273, 16905–16912. [DOI] [PubMed] [Google Scholar]
- Kido, S. , Kuriwaka-Kido, R. , Umino-Miyatani, Y. , Endo, I. , Inoue, D. , Taniguchi, H. , Inoue, Y. , Imamura, T. , Matsumoto, T. , 2010. Mechanical stress activates Smad pathway through PKCdelta to enhance interleukin-11 gene transcription in osteoblasts. PLoS One. 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler, R. , Distler, A. , Hoyer, J. , 1998. Pressure-activated cation channel in intact rat endocardial endothelium. Cardiovasc. Res. 38, 433–440. [DOI] [PubMed] [Google Scholar]
- Kovalenko, P.L. , Flanigan, T.L. , Chaturvedi, L. , Basson, M.D. , 2012. Influence of defunctionalization and mechanical forces on intestinal epithelial wound healing. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G1134–G1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraichely, R.E. , Farrugia, G. , 2007. Mechanosensitive ion channels in interstitial cells of Cajal and smooth muscle of the gastrointestinal tract. Neurogastroenterol. Motil. 19, 245–252. [DOI] [PubMed] [Google Scholar]
- Kumar, A. , Boriek, A.M. , 2003. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 17, 386–396. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Weaver, V.M. , 2009. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 28, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C.H. , Jeon, Y.T. , Kim, S.H. , Song, Y.S. , 2007. NF-kappaB as a potential molecular target for cancer therapy. Biofactors. 29, 19–35. [DOI] [PubMed] [Google Scholar]
- Lee, J.H. , Gomora, J.C. , Cribbs, L.L. , Perez-Reyes, E. , 1999. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys. J. 77, 3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemarie, C.A. , Esposito, B. , Tedgui, A. , Lehoux, S. , 2003. Pressure-induced vascular activation of nuclear factor-kappaB: role in cell survival. Circ. Res. 93, 207–212. [DOI] [PubMed] [Google Scholar]
- Less, J.R. , Posner, M.C. , Boucher, Y. , Borochovitz, D. , Wolmark, N. , Jain, R.K. , 1992. Interstitial hypertension in human breast and colorectal tumors. Cancer Res. 52, 6371–6374. [PubMed] [Google Scholar]
- Li, W. , Duzgun, A. , Sumpio, B.E. , Basson, M.D. , 2001. Integrin and FAK-mediated MAPK activation is required for cyclic strain mitogenic effects in Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 280, G75–G87. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Zou, L. , Zheng, Y. , Zhao, Z. , Li, Y. , Yang, P. , Luo, S. , 2007. NF-kappaB responds to mechanical strains in osteoblast-like cells, and lighter strains create an NF-kappaB response more readily. Cell Biol. Int. 31, 1220–1224. [DOI] [PubMed] [Google Scholar]
- Liu, M. , Xu, J. , Tanswell, A.K. , Post, M. , 1994. Inhibition of mechanical strain-induced fetal rat lung cell proliferation by gadolinium, a stretch-activated channel blocker. J. Cell Physiol. 161, 501–507. [DOI] [PubMed] [Google Scholar]
- Lu, F. , Chen, H. , Zhou, C. , Liu, S. , Guo, M. , Chen, P. , Zhuang, H. , Xie, D. , Wu, S. , 2008. T-type Ca2+ channel expression in human esophageal carcinomas: a functional role in proliferation. Cell Calcium. 43, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariot, P. , Vanoverberghe, K. , Lalevee, N. , Rossier, M.F. , Prevarskaya, N. , 2002. Overexpression of an alpha 1H (Cav3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J. Biol. Chem. 277, 10824–10833. [DOI] [PubMed] [Google Scholar]
- Martinac, B. , Adler, J. , Kung, C. , 1990. Mechanosensitive ion channels of E. coli activated by amphipaths. Nature. 348, 261–263. [DOI] [PubMed] [Google Scholar]
- Merritt, J.E. , Armstrong, W.P. , Benham, C.D. , Hallam, T.J. , Jacob, R. , Jaxa-Chamiec, A. , Leigh, B.K. , McCarthy, S.A. , Moores, K.E. , Rink, T.J. , 1990. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 271, 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic, M.F. , Pintilie, M. , Hedley, D.W. , Bristow, R.G. , Wouters, B.G. , Oza, A.M. , Laframboise, S. , Hill, R.P. , Fyles, A.W. , 2014. High tumor interstitial fluid pressure identifies cervical cancer patients with improved survival from radiotherapy plus cisplatin versus radiotherapy alone. Int. J. Cancer. 135, 1692–1699. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen, D. , Das, K. , Grimes, K.V. , 2012. Protein kinase C, an elusive therapeutic target?. Nat. Rev. Drug Discov. 11, 937–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourkioti, F. , Kratsios, P. , Luedde, T. , Song, Y.H. , Delafontaine, P. , Adami, R. , Parente, V. , Bottinelli, R. , Pasparakis, M. , Rosenthal, N. , 2006. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Invest. 116, 2945–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navalitloha, Y. , Schwartz, E.S. , Groothuis, E.N. , Allen, C.V. , Levy, R.M. , Groothuis, D.R. , 2006. Therapeutic implications of tumor interstitial fluid pressure in subcutaneous RG-2 tumors. Neuro Oncol. 8, 227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, C.M. , Jean, R.P. , Tan, J.L. , Liu, W.F. , Sniadecki, N.J. , Spector, A.A. , Chen, C.S. , 2005. Emergent patterns of growth controlled by multicellular form and mechanics. Proc. Natl. Acad. Sci. U S A. 102, 11594–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opsahl, L.R. , Webb, W.W. , 1994. Transduction of membrane tension by the ion channel alamethicin. Biophys. J. 66, 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek, M.J. , Zahir, N. , Johnson, K.R. , Lakins, J.N. , Rozenberg, G.I. , Gefen, A. , Reinhart-King, C.A. , Margulies, S.S. , Dembo, M. , Boettiger, D. , Hammer, D.A. , Weaver, V.M. , 2005. Tensional homeostasis and the malignant phenotype. Cancer Cell. 8, 241–254. [DOI] [PubMed] [Google Scholar]
- Peddibhotla, S. , Tepe, J.J. , 2004. Stereoselective synthesis of highly substituted Delta1-pyrrolines: exo-selective 1,3-dipolar cycloaddition reactions with azlactones. J. Am. Chem. Soc. 126, 12776–12777. [DOI] [PubMed] [Google Scholar]
- Peng, A. , Wu, T. , Zeng, C. , Rakheja, D. , Zhu, J. , Ye, T. , Hutcheson, J. , Vaziri, N.D. , Liu, Z. , Mohan, C. , Zhou, X.J. , 2011. Adverse effects of simulated hyper- and hypo-phosphatemia on endothelial cell function and viability. PLoS One. 6, e23268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju, B. , Haug, S.R. , Ibrahim, S.O. , Heyeraas, K.J. , 2008. High interstitial fluid pressure in rat tongue cancer is related to increased lymph vessel area, tumor size, invasiveness and decreased body weight. J. Oral Pathol. Med. 37, 137–144. [DOI] [PubMed] [Google Scholar]
- Sarntinoranont, M. , Rooney, F. , Ferrari, M. , 2003. Interstitial stress and fluid pressure within a growing tumor. Ann. Biomed. Eng. 31, 327–335. [DOI] [PubMed] [Google Scholar]
- Schwartz, E.A. , Bizios, R. , Medow, M.S. , Gerritsen, M.E. , 1999. Exposure of human vascular endothelial cells to sustained hydrostatic pressure stimulates proliferation. Involvement of the alphaV integrins. Circ. Res. 84, 315–322. [DOI] [PubMed] [Google Scholar]
- Sharma, V. , Lansdell, T.A. , Peddibhotla, S. , Tepe, J.J. , 2004. Sensitization of tumor cells toward chemotherapy: enhancing the efficacy of camptothecin with imidazolines. Chem. Biol. 11, 1689–1699. [DOI] [PubMed] [Google Scholar]
- Shiratsuch, H. , Basson, M.D. , 2005. Differential regulation of monocyte/macrophage cytokine production by pressure. Am. J. Surg. 190, 757–762. [DOI] [PubMed] [Google Scholar]
- Shiratsuchi, H. , Basson, M.D. , 2004. Extracellular pressure stimulates macrophage phagocytosis by inhibiting a pathway involving FAK and ERK. Am. J. Physiol. Cell Physiol. 286, C1358–C1366. [DOI] [PubMed] [Google Scholar]
- Sokabe, M. , Sachs, F. , Jing, Z.Q. , 1991. Quantitative video microscopy of patch clamped membranes stress, strain, capacitance, and stretch channel activation. Biophys. J. 59, 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer, K. , Guo, B. , Pomerantz, J.L. , Bandaranayake, A.D. , Moreno-Garcia, M.E. , Ovechkina, Y.L. , Rawlings, D.J. , 2005. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity. 23, 561–574. [DOI] [PubMed] [Google Scholar]
- Stark, L.A. , Reid, K. , Sansom, O.J. , Din, F.V. , Guichard, S. , Mayer, I. , Jodrell, D.I. , Clarke, A.R. , Dunlop, M.G. , 2007. Aspirin activates the NF-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis. 28, 968–976. [DOI] [PubMed] [Google Scholar]
- Sun, W. , Yang, J. , 2010. Molecular basis of lysophosphatidic acid-induced NF-kappaB activation. Cell Signal. 22, 1799–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, T. , Tamba, Y. , Masum, S.M. , Yamashita, Y. , Yamazaki, M. , 2002. La(3+) and Gd(3+) induce shape change of giant unilamellar vesicles of phosphatidylcholine. Biochim. Biophys. Acta. 1564, 173–182. [DOI] [PubMed] [Google Scholar]
- Thamilselvan, V. , Basson, M.D. , 2004. Pressure activates colon cancer cell adhesion by inside-out focal adhesion complex and actin cytoskeletal signaling. Gastroenterology. 126, 8–18. [DOI] [PubMed] [Google Scholar]
- Traub, O. , Monia, B.P. , Dean, N.M. , Berk, B.C. , 1997. PKC-epsilon is required for mechano-sensitive activation of ERK1/2 in endothelial cells. J. Biol. Chem. 272, 31251–31257. [DOI] [PubMed] [Google Scholar]
- Walsh, M.F. , Woo, R.K. , Gomez, R. , Basson, M.D. , 2004. Extracellular pressure stimulates colon cancer cell proliferation via a mechanism requiring PKC and tyrosine kinase signals. Cell Prolif. 37, 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J.S. , Pavlotsky, N. , Tauber, A.I. , Zaner, K.S. , 1993. Assembly dynamics of actin in adherent human neutrophils. Cell Motil. Cytoskeleton. 26, 340–348. [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y. , Gaynor, R.B. , 2001. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Invest. 107, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X.C. , Sachs, F. , 1989. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science. 243, 1068–1071. [DOI] [PubMed] [Google Scholar]