

Abstract
Background
Sturge-Weber syndrome (SWS) is a neurovascular disorder with a capillary malformation of the face (port-wine birthmark), a capillary-venous malformation in the eye, and a capillary-venous malformation in the brain (leptomeningeal angioma). Although SWS is a congenital disorder usually presenting in infancy, occasionally neurologic symptoms first present in adulthood and most affected individuals do survive into adulthood with varying degrees of neurologic impairment including epilepsy, hemiparesis, visual field deficits and cognitive impairments ranging from mild learning disabilities to severe deficits. SWS is a multi-system disorder that requires the neurologist to be aware of the possible endocrine, psychiatric, ophthalmologic, and other medical issues that can arise and impact the neurologic status of these patients. Some of these clinical features have only recently been described.
Review Summary
This review summarizes the neurologic manifestations of SWS, discusses issues related to the diagnosis of brain involvement, relates major neuroimaging findings, briefly describes the current understanding of pathogenesis, and provides an overview of neurologic treatment strategies.
Conclusion
Recent clinical research has highlighted several novel and lesser known aspects of this clinical syndrome including endocrine disorders. Functional imaging studies and clinical experience suggests that neurologic progression results primarily from impaired blood flow and that prolonged seizures may contribute to this process. Treatment is largely symptomatic although aggressive efforts to prevent seizures and strokes, in young children especially, may impact outcome.
Keywords: Sturge-Weber syndrome, port-wine birthmark, seizures, migraines, stroke
Sturge-Weber syndrome (SWS) is a congenital capillary-venous vascular malformation involving the brain (leptomeningeal angioma), skin (capillary malformation) and eye.1 The neurologic manifestations of SWS frequently evolve over the lifetime of the individual. While this process has been best studied in infants and young children with SWS, it is clear that patients can also present with new neurologic symptoms in adulthood and more research is also needed in this area. In the last decade, advanced neuroimaging and several clinical cohort studies of patients with Sturge-Weber syndrome have brought into focus a greater understanding of the range of clinical manifestations, improved our understanding of neurologic deterioration, and suggested promising new avenues for early diagnosis and treatment.
Neurologic Signs and Symptoms in Sturge-Weber Syndrome
Most commonly patients with brain involvement will present with a seizure in infancy. Typically seizures are focal motor seizures which can be very subtle in infants; complex partial seizures are also common. A small but significant subgroup will also have atonic seizures, and carbamazepine or oxcarbazepine can make these worse. 2,3 Infantile spasms are rare but have been reported and are usually asymmetric. 4 Status epilepticus is not uncommon and is frequently associated, with prolonged weakness on one side of the body or new onset visual-field deficit (lasting several days, weeks, or months), referred to as a stroke-like episode.5 Clusters of seizures, separated by many months or years of seizure freedom, are also common.3 In most cases, epilepsy begins in infancy or young childhood; however seizures can occasionally begin in adulthood and be the presenting symptom in an otherwise neurologically intact individual.6 Seizures occur in roughly 75% of SWS patients with unilateral brain involvement and in approximately 95% of patients with bilateral brain involvement.7,8 Rarely, instead of presenting with acute seizures, infants with SWS brain involvement present with early handedness, evidence of hemiparesis, or a visual gaze preference.
Neurologic deficits can be acquired slowly over time, or as the result of stroke-like episodes associated with seizures and/or migraines.1 Toddlers and young children are particularly prone to stroke-like episodes triggered by falls with minor head injury.9 Because of this propensity, patients with SWS should be advised to avoid recreational activities that are expected to frequently involve blows to the head. Seizure onset at less than six months of age has been related to increased severity of hemiparesis in a cohort of 77 patients seen at one center (Kossoff et al, 2009 [need numeric reference]). Most adults with SWS brain involvement have some degree of focal neurologic deficit, usually hemparesis.10 Neurologic deterioration is more common in infants and young children, but strokes and acquisition of new neurologic deficits can also occur in adults. Early onset dementia in the 5th or 6th decade can also occur but is poorly understood (clinical observation).
Cognitive impairments are common in patients with SWS but range greatly from attention problems or mild learning disability to severe cognitive impairment. Early onset of hard to control seizures and bilateral brain involvement may predict for more severe cognitive impairments.11 Neuropsychological testing starting at the age of 3 or 4 years can be helpful in identifying at-risk children. A recent study indicated that children with SWS and hemiparesis are likely to have global functional impairments, suggesting that hemiparesis on neurologic exam marks the young child who will likely benefit from neuropsychological testing even in the absence of obvious delays.12 Attention problems are responsive to stimulants (unpublished observations) which should be considered where suitable. Aggression and self-abusive behaviors are frequent in intellectually impaired adults with SWS. Depression is common in adults with SWS, particularly in those who are intellectually normal. 10
Headaches and migraines are common in SWS and, in adults, can more severely impact their quality of life than do their seizures.13 In patients with SWS, migraines frequently precede seizures; likewise seizures are frequently followed by headaches. Both seizures and migraines can be accompanied by a stroke-like episode. A recent report of an adult with SWS during a episode of intractable headache with prolonged visual aura highlighted focal hyperemia on SPECT, increased extent of occipital sulcal enhancement on MRI with contrast, and occipital retention of contrast on CTA during the headache. These findings resolved completely after the event and suggested a component of venous stasis.14
Miller et al. reported an increased prevalence of growth hormone deficiency in SWS (18 fold greater than the general population).15 Neuroimaging did not show apparent abnormalities involving the hypothalamic-pituitary axis and the cause of this deficiency remains uncertain. Nevertheless, growth hormone deficiency is a treatable cause of short stature or growth failure in patients with SWS. Very short adults with SWS should be evaluated for growth hormone deficiency, as this condition left untreated in adults can result in depression and medical problems.
Furthermore, central hypothyroidism associated with SWS has also been reported.16 The etiology of the central hypothyroidism is also not entirely clear. All children with SWS who have been diagnosed by us with central hypothyroidism (persistently low free T4 and low normal TSH) have been on chronic anticonvulsants. Central hypothyroidism (both symptomatic and asymptomatic) has been associated with the use of several of the anticonvulsants.17-19 However, given the increased risk of growth hormone deficiency in SWS, disruption of the hypothalamic-pituitary axis could also be a contributor to the central hypothyroidism issue.
Diagnosis of Sturge-Weber Sydrome Brain Involvement
SWS brain involvement should be suspected in any newborn with a port-wine birthmark (PWB, aka port-wine stain) in the V1 distribution (forehead to one side and/or upper eyelid). A PWB of any size in that location has approximately a 10-20% risk of SWS brain involvement. However the risk increases with the size, extent and bilaterality of the birthmark.20,21 Approximately 10% of those with typical SWS brain involvement will have no birthmark or eye involvement. These patients usually present with focal seizures later in childhood or adulthood and generally respond well to medical management. Unless the MRI is done with contrast, however, this diagnosis will be missed.22
SWS brain involvement is diagnosed by visualization of the typical leptomeningeal vascular malformation on contrast enhanced T1-weighted MRI imaging (Figure 1). Associated dilation and enhancement of the choroid plexus (glomus) on the involved side is also often present In older children and adults, dilated deep draining venous vessels underlying the effected cortical region may also be seen. Cortical and subcortical calcification is best seen on head CT (Figure 2).
Figure 1.
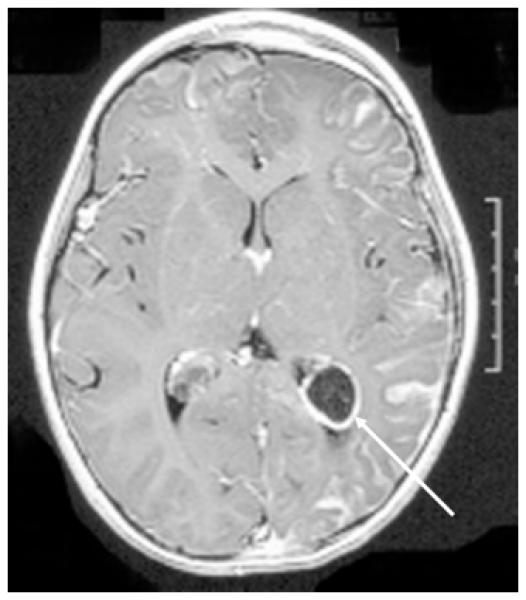
Post-contrast T1-weighted MRI of the brain demonstrating left hemispheric leptomeningeal angiomatosis and left choroid plexus enlargement and enhancement (glomus, see arrow).
Figure 2.
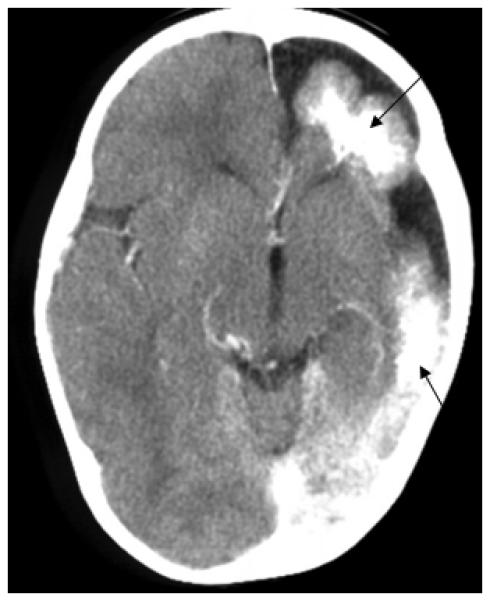
Head CT demonstrating significant left sided atrophy and cortical, sub-cortical calcification.
Early presymptomatic diagnosis of SWS brain involvement, however, is difficult because standard MRI and CT imaging have low sensitivity in the newborn period and early infancy. A negative head CT and/or contrast enhanced MRI of the brain in a normally developing asymptomatic young infant with a facial PWB does not exclude SWS brain involvement. The optimal timing of the first neuroimaging is unknown but current clinical experience suggests that if the child is developing normally, has a normal neurologic exam, no history of seizures, and a normal MRI with contrast after the age of 1 year, that child probably does not have SWS brain involvement. Post-contrast FLAIR and susceptibility-weighted MR imaging may provide additional sensitivity for involved brain areas. 23,24
Quantitative EEG (qEEG) is a promising new screening strategy currently being developed to select the most appropriate at-risk infants for neuroimaging. A decrease in qEEG power on the affected side correlates with the severity of neurologic deficits.25 Ewen et al. recently described a quantitative EEG approach that separated a group of nine infants with facial port-wine birthmark into affected and unaffected groups.26 If successfully applied to larger numbers of infants and EEGs from other facilities, this approach may prove to be a very useful tool for screening at-risk infants to determine who should receive an early MRI with contrast and which parents can be reassured and wait for later imaging.
Insights from Functional Imaging
The vascular malformation of the brain consists of hypoplastic cortical vessels associated with enlarged, tortuous leptomeningeal vessels and, frequently, with dilated deep venous vessels. Perfusion imaging suggests that impaired venous drainage from the involved brain regions results in impaired arterial perfusion.27 Eleven SPECT studies in young infants with SWS demonstrated that cerebral perfusion declines from being generous in the very young infant to being deficient in the involved cortical regions by the end of the first year of life.28 One reason that MRI with contrast is insensitive in newborns may be that the venous stasis and impaired blood flow dynamics have not yet developed. Two studies have reported ictal SPECT in a total of four individuals with SWS and showed that, during seizures, reduced increases in blood flow to the seizing hemisphere develop, or an absolute reduction in blood flow to ischemic levels ocurrs in the involved or remote brain areas.29,30 This finding suggests an abnormal vascular response to the seizures in SWS which likely further predisposes these patients to ischemic brain injury.
The question of whether seizures themselves exacerbate brain injury in SWS or are just a manifestation of the ischemia-induced brain injury remains controversial. When an infant or young child with SWS seizes for a prolonged period (status), they commonly have new or greatly worsened hemiparesis, new visual-field cut, and may regress in language and other cognitive milestones as well. This is often more than a Todd’s paralysis, as it may not fully resolve within a day, though it can improve or resolve over weeks and months if the child is subsequently free of seizures and stroke-like episodes. Given the ictal SPECT finding described above, abnormal autoregulation of blood flow during a long seizure or bout of multiple seizures is likely one mechanism for how seizures can exacerbate brain injury in SWS. This information argues for the aggressive management of seizures in SWS, especially early in the course of the disease.
Eventually PET imaging demonstrates decreased glucose metabolism in affected regions and can be useful in planning surgical resection by enhancing detection of abnormal brain regions. In most cases, seizures ensue, and when uncontrolled in young children, are associated with further deterioration in glucose metabolism over time in the affected brain regions. On the other hand, if seizures are well controlled for a prolonged period of time, glucose metabolism on PET imaging may improve.31 These PET imaging findings concur with the clinical observation that children whose seizures are well controlled generally make better developmental progress and their hemiparesis and visual-field deficit will often improve as well.
Pathogenesis of Sturge-Weber Syndrome and Neurologic Progression
A somatic mutation or double-hit mechanism for the etiology of SWS has been proposed,32 since the vascular malformation in SWS is usually localized to one region on one side of the body. During first trimester fetal development, the primitive vascular plexus invades the developing brain, skin, and eye, and a somatic mutation could derail the normal vascular maturation process. An alternative hypothesis proposed by Dr. Parsa suggested that all the main features of SWS arise from a localized primary venous dysplasia in the brain, with venous hypertension transmitted to the overlying skin and eye via persisting communicating collateral venous vessels.33 This putative brain venous dysplasia could itself be caused by a somatic mutation, however this hypothesis does not well explain individuals with extensive bilateral facial port-wine birthmarks and no evidence of SWS brain involvement at several years of age (clinical observation). The SWS vascular anomalies could result also from an environmental insult, however to date no maternal risk factor has been identified for the development of SWS. Nevertheless, in the effort to eventually create a plausible animal model of SWS, or to someday discover a strategy for preventing it altogether, both of these possible causative mechanisms need to be considered.
Microscopic examination of SWS brain tissue shows deposition of calcium in the cortex, hypoplastic blood vessels, gliosis, and sometimes loss of neurons or focal cortical dysgenesis. The meningeal vessels are enlarged and increased in number. Anoxic injury to endothelial cells or perithelial cells may contribute to the calcification. However, enhancement of the leptomeningeal vessels and enlarged deep venous vessels suggests breakdown of the blood–brain barrier, and calcification could therefore also result from increased blood vessel permeability.34 Port-wine birthmark blood vessels lack perivascular nerves,35,36 and SWS cortical vessels are innervated only by noradrenergic sympathetic nerve fibers.37 Increased endothelin-1 expression was found in SWS vascular malformations.38 Together these studies suggest that there may be increased vasconstrictive tone in the SWS brain vascular malformation; however, it is not known whether these alterations are pathologic or compensatory. Leptomeningeal vessels in SWS overexpress fibronectin,39 VEGF, the VEGF-receptor, and HIF-1.40 Increased endothelial proliferation and apoptosis are also reported. Therefore, the abnormal leptomeningeal blood vessels show evidence of ongoing vascular remodeling rather than being static lesions. However, this remodeling may provide a compensatory mechanism to maintain blood flow and therefore, at this time, it is unadvisable to try treatment of SWS brain involvement with anti-angiogenesis factors.
Treatment of Neurologic Symptoms
Since prolonged and frequent seizures appear to have a contributing role in the progression of neurologic injury, particularly in infants and young children. Therefore, educating parents on how to recognize and promptly treat seizures in SWS is important. Typically, the seizures in infants are subtle with rhythmic and persistant twitching of the hand or foot or deviation or nystagmus of the eyes. If the parents are expecting seizures to manifest with generalized tonic-clonic activity they may watch focal activity for hours without seeking medical attention.
I provide rectal diazepam for infants older than 3 months of age, even before definite brain involvement has been diagnosed in at-risk infants (since seizures may present before diagnosis can firmly be established or excluded). Working with the parents and the local hospital, a plan should be made for the child to be loaded with phosphenytoin as soon as possible and a chronic anticonvulsant initiated after the first focal seizure, as additional seizures invariably follow. Since bouts of seizures often occur during periods of illness, and venous stasis also has a role in strokes and stroke-like episodes, aggressive hydration, fever treatment, and appropriate treatment of any infections are also important. Likewise, patients and family members should, when able, receive the flu shot annually. It also make sense that iron deficiency and other causes of anemia, when present, should be diagnosed and treated.
Since venous stasis and microvascular thrombosis probably contribute to neurologic decline in SWS, daily anti-platelet, low-dose aspirin (3-5 mg/kg/day) has been used empirically in children with SWS.41 One small, open-label study found a decrease in the number of stroke-like episodes in patients with SWS receiving low-dose aspirin.5 Udani et al. reported on 6 subjects with SWS where the initiation of low-dose aspirin was associated with a decrease in seizures.42 Low-dose aspirin can be started at the onset of seizures or other focal neurologic deficits; alternatively some parents and physicians decide to start this as soon as pre-symptomatic brain involvement is established on imaging. Larger prospective open-label studies are currently under-way. Other forms of anticoagulation have yet to be studied in SWS, and therefore no information is available on which to base recommendations.
Chronic anticonvulsant use is central to the neurologic treatment of the majority of children and adults with SWS who have epilepsy. Oxcarbazepine is an obvious first-line medication; however, we recently described patients with SWS who were on oxcarbazepine and developed evidence of central hypothyroidism associated with symptoms that improved after treatment with levothyroxine.16 As discussed previously, this issue may arise secondary to the use of some anticonvulsants and has been described in association with oxcarbazepine. One study in 18 girls with epilepsy treated with oxcarbazepine showed that 67% developed asymptomatic central hypothyroidism that resolved when their medication was changed.18 Another report noted symptomatic central hypothyroidism in three patients with epilepsy that were treated either with levothyroxine (2 cases) or changing their AED.17 However, this issue has subsequently raised a significant treatment dilemma. Persistant central hypothyroidism associated with symptoms such as constipation, excessive sleepiness, decline in attention and cognitive skills, excessive weight gain, and poor vertical growth has now been diagnosed in several children with SWS on oxcarbazepine (clinical observation). It is difficult to determine whether these symptoms are due to the central hypothyroidism, the natural history of the SWS itself, or some other side effect of their medications. Other AEDs that can be considered as alternative or first-line AEDS are leviteracitam and topiramate. When the child’s seizures are well controlled, however, the parents are frequently reluctant to switch them to another AED. A trial of levothyroxine may then be tried to determine whether symptoms improve and hypothyroidism resolves.
The older child with SWS tends to stabilize1,31 and if doing reasonably well can be expected to continue to do so. However, adolescence can be another time of instability with regards to seizure control, with seizures sometimes restarting in individuals who have been seizure-free for years. If the seizures in an older child or adult are extremely prolonged (several hours) or associated with severe illness, a stroke with new permanent deficits can still result, however this is uncommon. Nevertheless the adolescent and young adult should be strongly cautioned against excessive and risky behaviors that would greatly increase their risk of severe seizures. Clinical experience and a recent survey 43 suggests that sleep deprivation is a common trigger for both migraines and seizures in this group and firm education regarding this fact can help bring seizures under control in some cases.
Despite several series demonstrating that surgical resection is effective, and although early hemispherectomy has been recommended, there is still debate about patient selection and the timing of surgery.44-47 Surgery should not be considered at the time of diagnosis since the agreed upon indication is medically refractory seizures. Those patients who have daily or weekly seizures associated with hemiparesis, visual field deficits, and significant developmental delays despite adequate trials of at least a couple of anticonvulsants (and arguably also low-dose aspirin) are very reasonable surgical candidates. The most difficult patients to advise are those with infrequent, but significant, seizure clusters separated by many months of seizure freedom and good developmental gains, and those patients with no or only mild neurologic deficits. Surgery should only be done at a center with a pediatric epilepsy surgery program; despite surgical improvements and growing experience, there is risk from intracranial hemorrhage and infection in patients with SWS. Long-term seizure freedom is clearly associated with complete resection of the involved brain regions,46,47 however this approach may increase the risk of morbidity. Vision is a particularly important issue, since surgery frequently results in a visual field deficit and glaucoma may also threaten vision in the affected eye(s); therefore overall risks to vision is a serious consideration. Hemispherectomy can be considered for children with bilateral brain involvement when monitoring establishes that most of the seizures are clearly coming from one hemisphere.48 Vagal nerve stimulator has occasionally been used in this population but little data is available to guide its use; results we have seen are consistent with the experience in other epilepsy populations.
Headaches and migraines in SWS are treated with standard abortive medications and, when frequent or severe, with preventative medications. Personal triggers for migraines should be sought and behavior modified where possible. A survey of 74 subjects with SWS and migraine reported that 35% used headache preventative medication.43 Preventative medications effective in reducing both seizures and migraines, such as topiramate, valproate and gabapentin, should be considered first. We also have experience with verapamil and periactin and have found them useful. The survey also reported that 22% of patients had received triptans and many experienced improvement; no strokes, hemorrhages or other serious complications were reported.43 Although use of triptans appears to be safe, the individual response to triptans should be carefully monitored. In many patients, headaches precede their stroke-like episode or seizure event and in these patients especially, every effort should be made to aggressively treat and prevent their headaches.
CONCLUSIONS
The tremendous variability seen in SWS greatly complicates the ability of the medical practitioner to advise a patient or family, particularly when the patient is young. Many important questions are still in great need of future study; some of these questions deal directly with adults with SWS, such as pregnancy and SWS and how the process of aging impacts the neurologic status of individuals with SWS. However, we now have a better understanding of the mechanisms of neurologic progression in SWS and new tools are being developed to aid early diagnosis. With the newer anticonvulsants, better migraine treatment, and low-dose aspirin, the aggressive medical management of SWS may now produce better results than previously. However this approach requires continued study as does the long term outcome of surgery in this population. The endocrine aspects of SWS are now being appreciated; this both complicates their care and may provide new opportunities to help these patients. Multi-centered efforts are being established that hold promise for new insights and treatment options in the future.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Bodensteiner John B., Roach ES. Sturge-Weber syndrome: Introduction and Overview. John B.Bodensteiner and E.S.Roach; 1999. pp. 1–10. [Google Scholar]
- 2.Ewen JB, Comi AM, Kossoff EH. Myoclonic-astatic epilepsy in a child with sturge-weber syndrome. Pediatr Neurol. 2007;36:115–117. doi: 10.1016/j.pediatrneurol.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Kossoff EH, Ferenc L, Comi AM. An infantile-onset, severe, yet sporadic seizure pattern is common in Sturge-Weber syndrome. Epilepsia. 2009;50:2154–2157. doi: 10.1111/j.1528-1167.2009.02072.x. [DOI] [PubMed] [Google Scholar]
- 4.Fukuyama Y, Tsuchiya S. A study on Sturge-Weber syndrome. Report of a case associated with infantile spasms and electroencephalographic evolution in five cases. Eur Neurol. 1979;18:194–204. doi: 10.1159/000115076. [DOI] [PubMed] [Google Scholar]
- 5.Maria BL, Neufeld JA, Rosainz LC, et al. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. J Child Neurol. 1998;13:606–618. doi: 10.1177/088307389801301204. [DOI] [PubMed] [Google Scholar]
- 6.Jacobs J, Levan P, Olivier A, Andermann F, Dubeau F. Late-onset epilepsy in a surgically-treated Sturge-Weber patient. Epileptic Disord. 2008;10:312–318. doi: 10.1684/epd.2008.0226. [DOI] [PubMed] [Google Scholar]
- 7.Bebin EM, Gomez MR. Prognosis in Sturge-Weber disease: comparison of unihemispheric and bihemispheric involvement. J Child Neurol. 1988;3:181–184. doi: 10.1177/088307388800300306. [DOI] [PubMed] [Google Scholar]
- 8.Oakes WJ. The natural history of patients with the Sturge-Weber syndrome. Pediatr Neurosurg. 1992;18:287–290. doi: 10.1159/000120677. [DOI] [PubMed] [Google Scholar]
- 9.Zolkipli Z, Aylett S, Rankin PM, Neville BG. Transient exacerbation of hemiplegia following minor head trauma in Sturge-Weber syndrome. Dev Med Child Neurol. 2007;49:697–699. doi: 10.1111/j.1469-8749.2007.00697.x. [DOI] [PubMed] [Google Scholar]
- 10.Sujansky E, Conradi S. Outcome of Sturge-Weber syndrome in 52 adults. Am J Med Genet. 1995;57:35–45. doi: 10.1002/ajmg.1320570110. [DOI] [PubMed] [Google Scholar]
- 11.Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49–58. doi: 10.1177/088307389501000113. [DOI] [PubMed] [Google Scholar]
- 12.Reesman J, Gray R, Suskauer SJ, et al. Hemiparesis Is a Clinical Correlate of General Adaptive Dysfunction in Children and Adolescents With Sturge-Weber Syndrome. J Child Neurol. 2009 doi: 10.1177/0883073808329529. [DOI] [PubMed] [Google Scholar]
- 13.Kossoff EH, Hatfield LA, Ball KL, et al. Comorbidity of epilepsy and headache in patients with Sturge-Weber syndrome. J Child Neurol. 2005;20:678–682. doi: 10.1177/08830738050200080901. [DOI] [PubMed] [Google Scholar]
- 14.Iizuka T, Sakai F, Yamakawa K, et al. Vasogenic leakage and the mechanism of migraine with prolonged aura in Sturge-Weber syndrome. Cephalalgia. 2004;24:767–770. doi: 10.1111/j.1468-2982.2004.00769.x. [DOI] [PubMed] [Google Scholar]
- 15.Miller RS, Ball KL, Comi AM, et al. Growth hormone deficiency in Sturge-Weber syndrome. Arch Dis Child. 2006;91:340–341. doi: 10.1136/adc.2005.082578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comi AM, Bellamkonda S, Ferenc LM, et al. Central hypothyroidism and Sturge-Weber syndrome. Pediatr Neurol. 2008;39:58–62. doi: 10.1016/j.pediatrneurol.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 17.Miller J, Carney P. Central hypothyroidism with oxcarbazepine therapy. Pediatr Neurol. 2006;34:242–244. doi: 10.1016/j.pediatrneurol.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 18.Vainionpaa LK, Mikkonen K, Rattya J, et al. Thyroid function in girls with epilepsy with carbamazepine, oxcarbazepine, or valproate monotherapy and after withdrawal of medication. Epilepsia. 2004;45:197–203. doi: 10.1111/j.0013-9580.2004.26003.x. [DOI] [PubMed] [Google Scholar]
- 19.Hirfanoglu T, Serdaroglu A, Camurdan O, et al. Thyroid function and volume in epileptic children using carbamazepine, oxcarbazepine and valproate. Pediatr Int. 2007;49:822–826. doi: 10.1111/j.1442-200X.2007.02456.x. [DOI] [PubMed] [Google Scholar]
- 20.Tallman B, Tan OT, Morelli JG, et al. Location of port-wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics. 1991;87:323–327. [PubMed] [Google Scholar]
- 21.Ch'ng S, Tan ST. Facial port-wine stains - clinical stratification and risks of neuro-ocular involvement. J Plast Reconstr Aesthet Surg. 2008;61:889–893. doi: 10.1016/j.bjps.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Comi AM, Fischer R, Kossoff EH. Encephalofacial angiomatosis sparing the occipital lobe and without facial nevus: on the spectrum of Sturge-Weber syndrome variants? J Child Neurol. 2003;18:35–38. doi: 10.1177/08830738030180010601. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths PD, Coley SC, Romanowski CA, et al. Contrast-enhanced fluid-attenuated inversion recovery imaging for leptomeningeal disease in children. AJNR Am J Neuroradiol. 2003;24:719–723. [PMC free article] [PubMed] [Google Scholar]
- 24.Mentzel HJ, Dieckmann A, Fitzek C, et al. Early diagnosis of cerebral involvement in Sturge-Weber syndrome using high-resolution BOLD MR venography. Pediatr Radiol. 2005;35:85–90. doi: 10.1007/s00247-004-1333-2. [DOI] [PubMed] [Google Scholar]
- 25.Hatfield LA, Crone NE, Kossoff EH, et al. Quantitative EEG asymmetry correlates with clinical severity in unilateral Sturge-Weber syndrome. Epilepsia. 2007;48:191–195. doi: 10.1111/j.1528-1167.2006.00630.x. [DOI] [PubMed] [Google Scholar]
- 26.Ewen JB, Kossoff EH, Crone NE, et al. Use of quantitative EEG in infants with port-wine birthmark to assess for Sturge-Weber brain involvement. Clin Neurophysiol. 2009;120:1433–1440. doi: 10.1016/j.clinph.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin DD, Barker PB, Kraut MA, et al. Early characteristics of Sturge-Weber syndrome shown by perfusion MR imaging and proton MR spectroscopic imaging. AJNR Am J Neuroradiol. 2003;24:1912–1915. [PMC free article] [PubMed] [Google Scholar]
- 28.Pinton F, Chiron C, Enjolras O, et al. Early single photon emission computed tomography in Sturge-Weber syndrome. J Neurol Neurosurg Psychiatry. 1997;63:616–621. doi: 10.1136/jnnp.63.5.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Namer IJ, Battaglia F, Hirsch E, et al. Subtraction ictal SPECT co-registered to MRI (SISCOM) in Sturge-Weber syndrome. Clin Nucl Med. 2005;30:39–40. doi: 10.1097/00003072-200501000-00014. [DOI] [PubMed] [Google Scholar]
- 30.Aylett SE, Neville BG, Cross JH, et al. Sturge-Weber syndrome: cerebral haemodynamics during seizure activity. Dev Med Child Neurol. 1999;41:480–485. [PubMed] [Google Scholar]
- 31.Juhasz C, Batista CE, Chugani DC, et al. Evolution of cortical metabolic abnormalities and their clinical correlates in Sturge-Weber syndrome. Eur J Paediatr Neurol. 2007 doi: 10.1016/j.ejpn.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- 33.Parsa CF. Sturge-weber syndrome: a unified pathophysiologic mechanism. Curr Treat Options Neurol. 2008;10:47–54. doi: 10.1007/s11940-008-0006-0. [DOI] [PubMed] [Google Scholar]
- 34.Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509–516. doi: 10.1177/08830738030180080701. [DOI] [PubMed] [Google Scholar]
- 35.Rydh M, Malm M, Jernbeck J, et al. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419–422. doi: 10.1097/00006534-199103000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Smoller BR, Rosen S. Port-wine stains. A disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177–179. doi: 10.1001/archderm.122.2.177. [DOI] [PubMed] [Google Scholar]
- 37.Sa M, Barroso CP, Caldas MC, et al. Innervation pattern of malformative cortical vessels in Sturge-Weber disease: an histochemical, immunohistochemical, and ultrastructural study. Neurosurgery. 1997;41:872–876. doi: 10.1097/00006123-199710000-00020. [DOI] [PubMed] [Google Scholar]
- 38.Rhoten RL, Comair YG, Shedid D, et al. Specific repression of the preproendothelin-1 gene in intracranial arteriovenous malformations. J Neurosurg. 1997;86:101–108. doi: 10.3171/jns.1997.86.1.0101. [DOI] [PubMed] [Google Scholar]
- 39.Comi AM, Weisz CJ, Highet BH, et al. Sturge-Weber syndrome: Altered blood vessel fibronectin expression and morphology. J Child Neurol. doi: 10.1177/08830738050200070601. In press. [DOI] [PubMed] [Google Scholar]
- 40.Comati A, Beck H, Halliday W, et al. Upregulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha in leptomeningeal vascular malformations of Sturge-Weber syndrome. J Neuropathol Exp Neurol. 2007;66:86–97. doi: 10.1097/nen.0b013e31802d9011. [DOI] [PubMed] [Google Scholar]
- 41.Garcia JC, Roach ES, McLean WT. Recurrent thrombotic deterioration in the Sturge-Weber syndrome. Childs Brain. 1981;8:427–433. doi: 10.1159/000120011. [DOI] [PubMed] [Google Scholar]
- 42.Udani V, Pujar S, Munot P, et al. Natural history and magnetic resonance imaging follow-up in 9 Sturge-Weber Syndrome patients and clinical correlation. J Child Neurol. 2007;22:479–483. doi: 10.1177/0883073807300526. [DOI] [PubMed] [Google Scholar]
- 43.Kossoff EH, Balasta M, Hatfield LM, et al. Self-reported treatment patterns in patients with Sturge-Weber syndrome and migraines. J Child Neurol. 2007;22:720–726. doi: 10.1177/0883073807304008. [DOI] [PubMed] [Google Scholar]
- 44.Kossoff EH, Buck C, Freeman JM. Outcomes of 32 hemispherectomies for Sturge-Weber syndrome worldwide. Neurology. 2002;59:1735–1738. doi: 10.1212/01.wnl.0000035639.54567.5c. [DOI] [PubMed] [Google Scholar]
- 45.Steinbok P, Gan PY, Connolly MB, et al. Epilepsy surgery in the first 3 years of life: a Canadian survey. Epilepsia. 2009;50:1442–1449. doi: 10.1111/j.1528-1167.2008.01992.x. [DOI] [PubMed] [Google Scholar]
- 46.Arzimanoglou AA, Andermann F, Aicardi J, et al. Sturge-Weber syndrome: indications and results of surgery in 20 patients. Neurology. 2000;55:1472–1479. doi: 10.1212/wnl.55.10.1472. [DOI] [PubMed] [Google Scholar]
- 47.Bourgeois M, Crimmins DW, de Oliveira RS, et al. Surgical treatment of epilepsy in Sturge-Weber syndrome in children. J Neurosurg. 2007;106:20–28. doi: 10.3171/ped.2007.106.1.20. [DOI] [PubMed] [Google Scholar]
- 48.Tuxhorn IE, Pannek HW. Epilepsy surgery in bilateral Sturge-Weber syndrome. Pediatr Neurol. 2002;26:394–397. doi: 10.1016/s0887-8994(01)00414-3. [DOI] [PubMed] [Google Scholar]