

Abstract
Cofilin (CFL) is an F-actin–severing protein required for the cytoskeleton reorganization and filopodia formation, which drives cell migration. CFL binding and severing of F-actin is controlled by Ser3 phosphorylation, but the contributions of this step to cell migration during invasion and metastasis of cancer cells are unclear. In this study, we addressed the question in prostate cancer cells, including the response to TGF-β, a critical regulator of migration. In cells expressing wild-type CFL, TGF-β treatment increased LIMK-2 activity and cofilin phosphorylation, decreasing filopodia formation. Conversely, constitutively active CFL (SerAla) promoted filipodia formation and cell migration mediated by TGF-β. Notably, in cocultures of prostate cancer epithelial cells and cancer-associated fibroblasts, active CFL promoted invasive migration in response to TGF-β in the microenvironment. Further, constitutively active CFL elevated the metastatic ability of prostate cancer cells in vivo. We found that levels of active CFL correlated with metastasis in a mouse model of prostate tumor and that in human prostate cancer, CFL expression was increased significantly in metastatic tumors. Our findings show that the actin-severing protein CFL coordinates responses to TGF-β that are needed for invasive cancer migration and metastasis.
Introduction
One of six men will be diagnosed with prostate cancer during their lifetime. One out of 36 patients will not survive the disease. Death from prostate cancer results when cancer cells become metastatic after invading lymph nodes and blood vessels and migrate to the bone (1). The mainstay of treatment for metastatic prostate cancer has been androgen ablation, conferring improved survival to patients with advanced disease (2). The apoptotic response to androgen ablation is the underlying mechanism driving tumor regression and therapeutic benefit in patients with prostate cancer (3). The majority of prostate tumors recur, however, emerging as castration-resistant, causing mortality due to distinct molecular and genetic changes, reactivating the androgen receptor signaling, and conferring resistance to androgen deprivation–induced apoptosis (4, 5). Dynamic interactions between cancer cells and the tumor microenvironment consisting of the reactive stroma (6, 7), inflammatory cells, angiogenic vessels, fibroblasts, and components of the extracellular matrix (ECM), dictate tumor invasion (8-10) and confer androgen insensitivity in metastatic prostate cancer (11).
Cancer metastasis is regulated by signaling contributions from the microenvironment promoting cell detachment from the primary tumor and ECM (12, 13), acquisition of epithelial– mesenchymal transition (EMT) phenotype and invasion into the surrounding tissue (14), resistance to anoikis (15, 16), and migration via a chemoattractive path to a metastatic site (17, 18). A distinct molecular program regulates the adhesion, EMT, migratory, and invasive properties of disseminating tumor cells, all processes affected by the dynamics of the cytoskeleton (16, 19). The cellular cytoskeleton consists of a dynamic net of actin filaments, rapid polymerization and depolymerization, which allows the cell to move toward extracellular stimuli (20-22). The actin depolymerizing factor/cofilin protein is a small (19 kDa) actin-binding protein and the major regulator of the actin dynamics via the binding and severing of the filamentous actin form (23, 24). CFL binding causes a reduction in actin filament rotation allowing a free barbed end for the addition of new actin monomers (25, 26). CFL phosphorylation on Ser3 by LIMK-2 inhibits its binding to G actin (monomeric actin) and F-actin (filamentous actin) and severing of the actin filament in mammalian cells (19, 27). The Rho-associated, coiled-coil–containing protein kinase 1 (ROCK1) is responsible for LIMK-2 phosphorylation and activation (28, 29). Actin polymerization regulated by CFL dephosphorylation/activation is a convergence point in the intracellular network through which extracellular stimuli affect actin cytoskeleton, invasion, and apoptosis (21, 25, 30).
TGF-β functionally contributes to tumor progression to metastasis via its critical control of apoptosis and proliferation in the early stages of tumorigenesis, and its ability to promote angiogenesis, migration, invasion, and immunity in late stages of metastatic spread (31-33). TGF-β signaling is propagated via (serine threonine kinases) transmembrane receptors (TβRI and TβRII), activation of which leads to downstream regulation of SMADs intracellular effectors in target cells, including prostate cancer (34-36). The mechanism via which TGF-β is functionally converted from a suppressor of premalignant cells to a tumor progression supporter of metastasis is unknown (37). Elucidating the contribution of the actin cytoskeleton to this critical phenomenon in prostate tumorigenesis will enable a novel platform for defining predictive markers of metastasis, as well as potential therapeutic targets. CFL was previously identified in this laboratory as a Smad-independent intracellular effector of TGF-β signaling in prostate cancer cells (38). This study investigated the role of active CFL in directing TGF-β to elicite metastatic responses in prostate cancer. We generated a series of cofilin-phosphorylated/dephosphorylation mutants determing the actin severing activity and their impact on prostate tumor aggressive behavior was characterized in the context of the microenvironment in vitro and in vivo. Our findings support a functional contribution of active (dephosphorylated) cofilin to prostate cancer invasive and metastatic properties via switching TGF-β attributes.
Materials and Methods
Cell culture and transfections
The human prostate cancer cell lines PC-3 and LNCaP were obtained from the American Type Culture Collection. Cell lines were obtained every year between 2009 and 2013 and have been authenticated and tested for mycoplasma in August 2009, October 2010, May 2011, November 2012, and June 2013 by short tandem repeat (method of Masters et al. 2012: Authentication of human cell lines: standardization of STR profiling; DDC Medical). The LNCaP TβRII cells are stable transfectants established in our laboratory with restored sensitivity to TGF-β by overexpressing the human TβRII receptor (34). Primary cultures of human prostate cancer-associated fibroblasts (CAF) were established from radical prostatectomy specimens (from patients with prostate cancer) by Dr. S. Koochekpour (Roswell Park Cancer Institute, Buffalo, NY). All primary cultures were obtained through an Institutional Review Board– approved protocol in abidance with the guidelines at Roswell Park Cancer Institute (Buffalo, NY). Primary cultures of human prostate fibroblasts were passaged in the user’s laboratory not more than three times before use for the coculture experiments. The S3A and T25A cofilin-mutant prostate cancer cell lines were generated by site-directed mutagenesis in PC-3 cells. Briefly, a point mutation targeting the Ser 3 phosphorylation or the threonine 25 sites was induced by PCR. To mimic a dephosphorylated (constitutively active) form of cofilin (S3ACFL), a substitution of a serine on position 3 to alanine was generated. Wild-type (WT)-, S3ACFL-, and T25ACFLmutant forms of cofilin were introduced into PC-3 cells via stable transfection. S3D cofilin mutation, mimicking the constitutive phosphorylated (inactive) form, was introduced in PC-3 cells via transient transfection. Expression vectors for S3D cofilin were generously provided by Dr. Krupenko (Medical University of South Carolina, Charleston, SC; ref. 30). Cells were transfected with pXJN-HA/cofilin vector DNA using the Effectene Transfection Reagent (QIAGEN 301425). Cofilin expression was silenced by using the siRNA sequence targeting cofilin codons 64–84. An siRNA containing a two single-nucleotide mutation of cofilin (C71G and A73U) was used as control. CAFs are maintained in Stromal Cell Basel Medium (SCBM) CC-3204 (Lonza). Cultured cells were maintained at 30°C in a humidified 5% CO2 incubator. TGF-β ligand and its neutralizing antibody are from R&D Systems.
Cell viability evaluation
Cells (104 per well) cultured in 96-well plates are exposed to MTT reagent (4 hours) and after dissolving formazan crystals absorbance is read at 490nm(Bio-Rad 680).
Migration assays
Cells seeded in 6-well plates are wounded at 65% to 70% density of cell monolayers and exposed to TGF-β (5 ng/mL; 24 hours) in the presence or absence of TGF-β– neutralizing antibody. The number of migrating cells is counted in three different fields.
Adhesion assays
Cells are incubated in 6-well fibronectin-coated plates for 30 minutes; nonadherent cells are removed and adherent cells are fixed and evaluated under microscopy.
Cell invasion assays
Matrigel invasion assay
The invasion potential was evaluated using a Biocat Matrigel Transwell Chamber (Beckon Dickinson). Prostate cells were seeded into the upper chamber of a transwell insert precoated with fibronectin in serum-free medium (50,000 cells/well). After 24 hours, noninvading cells were removed from upper chamber and invading cells were stained with Diff-Quick Solution (IMEB Inc.).
Matrigel cell tracking
Human prostate CAFs (Supplementary Fig. S3) and PC-3 prostate cancer cells were independently grown in medium containing SCBM or RPMI, respectively. SCBM containing CellTracker Green CMFDA dye (5 μmol/L; Invitrogen) and 1640 RPMI medium containing CellTracker Orange CMTML (Invitrogen) were added to CAFs and PC-3 cells, respectively (45 minutes). The CellTracker Green CMFDA–labeled cell suspensions of CAFs were placed into the inner circle of underside membrane of a Biocat Matrigel Transwell Chamber. Inserts were placed in 12-well plates in Biocat Matrigel Transwell Chambers in the absence or presence of TGF-β ligand (5 ng/mL). CellTracker Orange CMTML–labeled WT and S3ACFL PC-3 cells were seeded in the upper chamber and after 24 hours invading cells were detected using an epifluorescence Nikon Eclipse E600 microscope (Nikon).
In vitro coculture assay
Human prostate CAFs (Supplementary Fig. S3) were grown in the inner membrane circle of Biocat Matrigel Transwell Chamber inserts and after 24 hours, inserts were transferred in Biocat Matrigel Transwell Chambers in the absence/presence of TGF-β–neutralizing antibody. Prostate cancer epithelial cells were seeded into the upper chamber and after coculturing for 24 hours, invading cells were stained with Diff-Quick Solution (IMEB Inc.).
Western blot and immunoprecipitation analysis
Cell pellets and lung tissue were lysed in radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl, pH7.4, 1% NP40, 0.25% Na-deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L phenylmethylsulfonylfluoride, (Sigma P8340 protease inhibitor). Cell lysates were subjected to SDS-PAGE and transferred to Immun-Blot PVDF membranes. After exposure to the respective primary antibodies, proteins were detected using the ECL Plus Detection System (GE). The antibodies used were anticofilin (Sigma-Aldrich), phospho-cofilin (Ser 3); anti-LIMK-2 (Cell Signaling Technology), and GFP (Santa Cruz Biotechnology). For the immunoprecipitation experiments, PC-3 cells were transfected with Flag-tagged WTCFL, S3ACFL, and T25A CFL, and cells were grown in charcoal-stripped serum medium for 24 hours. Cells were subsequently treated with TGF-β1 (for 6 hours) in the absence or presence of MAP–ERK kinase (MEK) inhibitor PD98095. Whole cell lysates were subjected to immunoprecipitation with the anti-Flag antibody, and Western blots with the specific antibodies.
Immunofluorescence analysis
Cells (7 × 104 cells/well) seeded in 6-well plates were exposed to TGF-β (5 ng/mL, 24 hours). Cells were fixed with methanol-free formaldehyde and permeabilized with Triton X-100 (0.1% v/v). Fluorescent staining of filamentous actin is performed using rhodamine phalloidin staining of F-actin (Invitrogen). Cofilin expression was detected using the rabbit anticofilin antibody following incubation with Alexa Fluor 488 (Invitrogen; 24 hours). Images were processed using a fluorescence Nikon Eclipse E600 microscope (Nikon).
Experimental metastasis assay
The metastatic potential of WTCFL- and S3ACFL-mutant PC-3 cells was examined in vivo by the tail vein injection– experimental metastasis assay. Male nude mice (6 weeks old; Harlan Laboratories Inc.) were maintained in sterile cages in pathogen-free environment. Animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee. GFP-labeled WTCFL and S3ACFL PC-3 cells (106) were injected into the tail vein of mice (n = 6/cell line). Four weeks after inoculation, lungs were excised and metastatic lesions to the lungs were examined under the microscope. Lung tissue was homogenized and subjected to Western blot analysis.
Immunohistochemical analysis
Human prostate specimens
Formalin-fixed paraffinembedded specimens of human prostate cancer, primary and metastatic (n = 11), were obtained from the Markey Biospecimen and Tissue Procurement Shared Resource Facility (BSTP SRF). Tissue sections (4 μm) were analyzed for cofilin and p-cofilin immunoreactivity, using antibodies cofilin (Sigma) and phospho-cofilin (Ser 3; Cell Signaling Technology). Palladin expression was detected using the palladin antibody (Proteintech Group Inc.). E-Cadherin was detected using the E-cadherin antibody (Cell Signaling Technology). H-scoring was assessed in three fields [cell positivity (Q) and intensity (I), graded as in Supplementary Fig. S5].
TRAMP mouse model
TRAMP transgenic mice develop prostate adenocarcinoma, resembling the clinical progression of human disease to metastasis (33). Tissue sections from TRAMP prostate tumors of increasingly aggressive stage were subjected to immunohistochemical analysis as described above.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 5 for Windows (GraphPad Software). Data are presented as means ±SEM. Numerical values are the mean of three independent experiments. Statistical evaluation of the data was performed using the Student t test and two-way ANOVA for multiple comparisons. Significant difference is defined at a P value of <0.05.
Results
Cofilin activity directs TGF-β–mediated actin severing in prostate cancer cells
Recent work on the actin cytoskeleton dynamics in prostate cancer metastasis led to the characterization of significant protein interactions (in the tumor microenvironment), targeting of which potentially impairs metastatic progression (39-42). The present study identified the functional contribution of CFL to the process of prostate cancer metastasis in the context of processing signals from the microenvironment. Previously, we identified CFL as a Smadindependent effector of TGF-β–mediated apoptosis signaling in prostate cancer cells by virtue of its cytosolic release (38). To assess the effect of exogenous TFG-β on CFL phosphorylation status and activity, constitutively active (dephosphorylated) forms of CFL were generated in PC-3 prostate cancer cells by mutagenesis via substitution of a serine on position 3 to alanine (S3ACFL) and a threonine to alanine at position 25 (T25ACFL). Immunoprecipitation analysis of phosphorylated protein associations in response to TGF-β revealed that the S3ACFL mutation specifically conferring CFL dephosphorylation promotes its association with actin (enhancing filament severing), whereas the T25ACFL mutation (impairing threonine phosphorylation) had no effect on the association of p-cofilin with actin (Fig. 1). Moreover, the presence of MEK inhibitor (PD98059) abrogated the TGF-β–mediated association between p-Erk and cofilin (Fig. 1). The mutational activation/cofilin dephosphorylation status (S3ACFL or T25ACFL) or silencing CFL expression (shCFL) had no significant consequences on prostate cancer cell viability (Supplementary Fig. S1A). TGF-β treatment significantly increased the WTCFL PC-3 cell invasion (inactive cofilin), whereas loss of cofilin abolished prostate cancer cell invasion (Supplementary Fig. S1B). PC-3 cells harboring the constitutive dephosphorylated (active) cofilin (S3ACFL) had a modestly enhanced invasive response to TGF-β (Supplementary Fig. S1B). The S3ACFL mutation, as expected, abrogated its phosphorylation by LIMK-2, without affecting total cofilin expression (Fig. 2A); there was a compensatory upregulation of LIMK-2 levels in the S3ACFL PC-3 cells compared withWTCFL (Fig. 2A). Figure 2B reveals the endogenous upregulation of downstream signaling effectors RhoA and ROCK1 induced by the introduction of cofilin mutations; both S3DCFL and S3ACFL cells exhibited a significant increase in protein expression for RhoA and ROCK1 compared with WTCFL cells. In response to TGF-β, there was a transient induction in phosphorylated CFL within 3 to 6 hours, which was preceded by a significant increase in ROCK 1 and Rho A levels in theWTCFL but not in the S3ACFL cells (Fig. 2C and D). This temporal phosphorylation of CFL in response to TGF-β was also detected in another human prostate cancer cell line (LNCaP TβRII) that has restored sensitivity to the cytokine (Supplementary Fig. S2).
Figure 1.
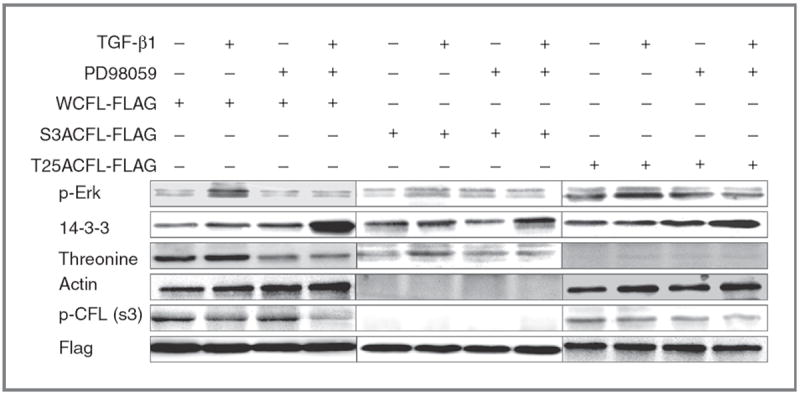
S3A cofilin dephosphorylation specifically directs actin severing response to TGF-β. PC-3 cells were transfected with Flag-tagged WTCFL, S3ACFL, or T25ACFL and were subsequently treated with TGF-β1 (5 ng/mL) for 6 hours in the presence or absence of PD98095 inhibitor. Cell lysates (50 μg of protein) were subjected to immunoprecipitation with anti-Flag antibody and subsequent Western blotting with the indicated antibodies. Phosphorylated proteins p-Erk, protein 14-3-3, actin, and p-cofilin are induced in response to TGF-β in WTFL prostate cancer cells. S3ACFL mutation conferring constitutive cofilin dephosphorylation specifically diminishes p-cofilin and enables active cofilin binding to actin without TGF-β regulation. In contrast, the T25A CFL mutation impairs threonine phosphorylation that does not affect actin binding, and thus both p-cofilin (Ser 3) and actin are detected.
Figure 2.
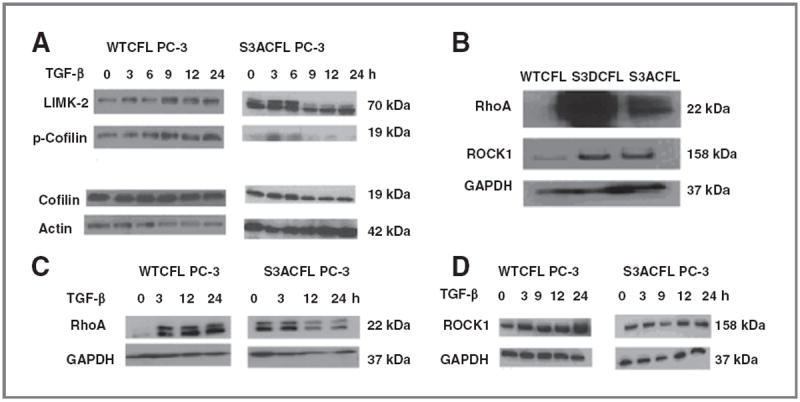
Effect of S3A mutation on cofilin phosphorylation events in PC-3 cells. Effect of TGF-β on cofilin, p-cofilin, and LIMK-2 protein expression in prostate cancer cells. A, upregulation of LIMK-2 protein in mutant S3ACFL PC-3 cells. Treatment with TGF-β (5 ng/mL) increased LIMK-2 and p-cofilin expression in the WT and decreased LIMK-2 expression in the S3ACFL cells. B, Western blotting indicating elevated RhoA and ROCK1 protein in S3ACFL PC-3. C and D, treatment with TGF-β increased RhoA and ROCK1 levels in WTCFL cells and decreased expression of both proteins in S3ACFL cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control.
Active cofilin dictates prostate cancer cell migration and invasion responses to TGF-β (via reactive stroma)
The effect of TGF-β on prostate cancer cell migration was analyzed in two different human prostate cancer cell lines, LNCaP TβRII and WTCFL PC-3, with normal CFL function. Treatment with TGF-β (24 hours) led to a significant decrease in prostate cancer cell migration for both cell lines (Fig. 3A and B). Functional blocking of TGF-β by the neutralizing antibody against the cytokine restored migration capacity to control levels in both cell lines LNCaP TβRII and WTCFL PC-3 (Fig. 3). To determine the effect of a mutation on CFL phosphorylation site in prostate cancer cell migration, we comparatively assessed the migration ability of S3ACFL mutants mimicking the constitutively active form of cofilin and, as a functional control, S3DCFL mimicking the constitutively inactive form. The S3A cofilinmutation resulted in a significant increase in PC-3 cell migration (Fig. 3B), whereas the induced loss of TGF-β (by neutralizing antibody) further increased S3ACFL cell migration (Fig. 3B). In contrast, a significant decrease in both cell migration (Supplementary Fig. S3A) and cell adhesion to ECM (Supplementary Fig. S3B) was observed for the inactive S3DCFL cells compared with the S3ACFL cells. The impact of S3ACFL mutation on prostate cancer cell invasion was interrogated in the context of tumor microenvironment. The quantitative data from the invasion assay indicate no significant difference in the invasion potential of S3ACFL cells compared with WTCFL PC-3 cells (Fig. 4A). The increase in invading cell number in response to exogenous TGF-β was abrogated by the presence of the neutralizing antibody against TGF-β, in both WTCFL and S3ACFL cells (P < 0.05).
Figure 3.
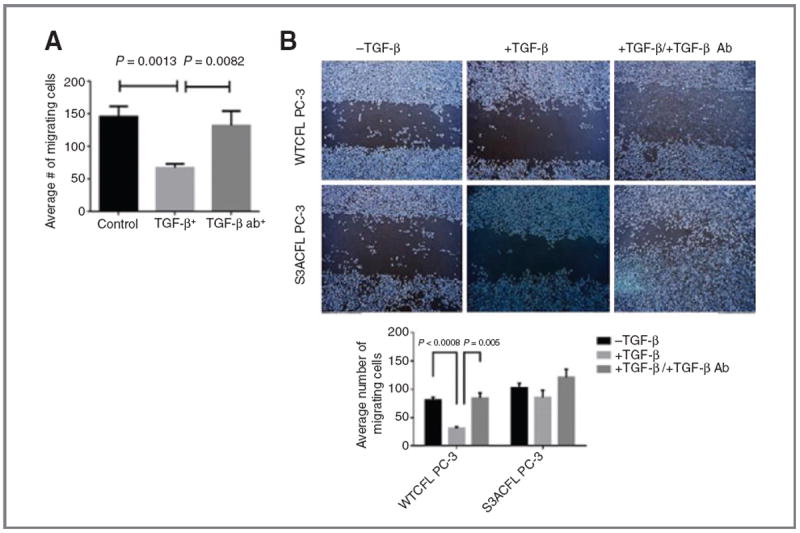
Cofilin drives TGF-β–mediated prostate cancer migration. A, human prostate cancer cells LNCaP TβRII (normal cofilin) exhibited a significant reduction in cell migration in response to TGF-β. In the presence of a neutralizing antibody against TGF-β, cell migration was restored to untreated control levels. Values shown are the number of migrating cells from two independent experiments performed in triplicate. B, S3ACFL mutation enhances prostate cancer cell migration bypassing TGF-β. Top, representative images of increased cell migration ability for S3ACFL PC-3 cells compared with WTCFL cells (24 hours). Bottom, TGF-β treatment significantly decreased WTCFL cell migration (P < 0.0008), but it had no significant effect in S3ACFL cells. Loss of TGF-β (by neutralizing antibody) restored the WTCFL PC-3 cell migration capacity, whereas it increased S3CFL-mutant cell migration (P = 0.005).
Figure 4.
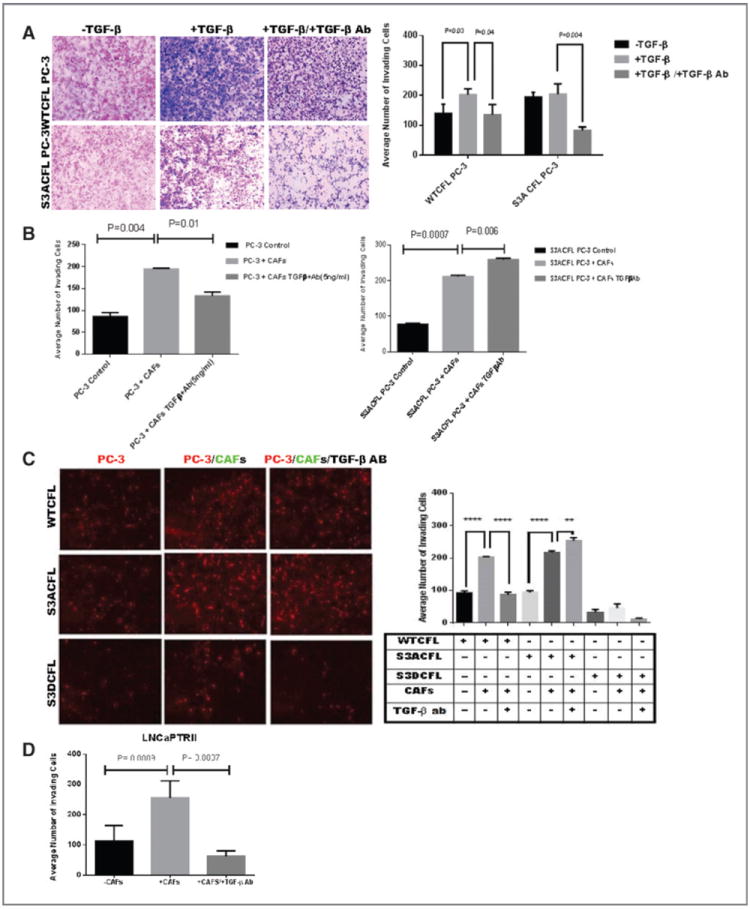
Cofilin navigates invasive response to TGF-β (from stroma microenvironment). A, the invasive response of prostate cancer cells to TGF-β assessed in the Matrigel assay. S3ACFL had no significant effect on PC-3 cell invasion (black barographs). In response to exogenous TGF-β, there was an increase in WTCFL PC-3 cell invasion potential, but not in S3ACFL cells (P = 0.03). In the presence of TGF-β–neutralizing antibody, there was a significant decrease in the invasion potential for both WTCFL and S3ACFL cells (P = 0.04 and 0.004, respectively). B, Matrigel invasion of WTCFL PC-3/CAFs (right) and S3ACFL PC-3/CAFs cocultures (left) after 24 hours. CAFs significantly increased prostate cancer cell invasion for both WTCFL and S3ACFL cells (P = 0.004 and 0.007. Continuous secretion of TGF-β by the reactive microenvironment (in the presence of TGF-β–neutralizing antibody) induced a further increase in the number of invading S3ACFL cells (P = 0.008), whereas it decreased WTCFL cell invasion. Values are the average from two independent experiments in triplicate. C, cocultures of WTCFL, S3ACFL, and S3DCFL with CAFs in the presence or absence of a neutralizing TGF-β antibody. Quantitative assessment of invading cells indicates that only active cofilin (S3A CFL) directs a further increase in TGF-β–mediated cell invasion (derived from CAFs). D, a significantly higher increase in cell invasion is stimulated by CAFs in cocultures with theLNCaP TβRII cells (highly responsive to TGF-β). This was abrogated by the presence of the neutralizing antibody against TGF-β. *, significant difference at P < 0.007.
The reactive stroma contributes to prostate cancer progression through the CAFs that facilitate metastasis (39, 40). To assess whether the effect of CFL on prostate tumor cell invasion is TGF-β dependent as mediated from surrounding CAFs (Supplementary Fig. S4), prostate cancer cell invasion was evaluated in in vitro cocultures. Fluorescent-labeled PC-3 prostate cancer epithelial cells (red) were cocultured with labeled human prostate CAFs (green) in the upper chamber of a Matrigel precoated transwell insert for 24 hours (Fig. 4B). Only prostate tumor epithelial cells invaded the Matrigel. There was no significant difference in cell invasion between WTCFL and S3ACFL cell lines (but there was a decrease in the S3D-mutant cells). In the presence of human prostate CAFs, however, there was a significant increase in the number of tumor epithelial cells invading for both WTCFL PC-3 and LNCaP TβRII cells (Fig. 4C and D, respectively). The S3D CFL mutation (phosphorylation) had no effect on prostate cancer cell invasion regardless of TGF-β status (Fig. 4C). Simultaneous exposure to the TGF-β–neutralizing antibody yielded an additional significant increase in the S3ACFL cell invasion potential (P < 0.004). In contrast, in two different prostate cancer cell lines with normal cofilin function WTCFL PC-3 cells and LNCaP TβRII cells, we observed the expected reduction in invasion. These data demonstrate that only active cofilin was able to functionally direct TGF-β signaling (secreted by the CAFs in the microenvironment) in promoting invasive behavior.
Cofilin mediates prostate cancer cell adhesion via cytoskeletal remodeling
Cell adhesion is directly dependent on cofilin activity and cytoskeletal actin because depolymerization and polymerization of new actin filaments is required for filopodia formation (41). We subsequently investigated the effect of S3A mutation on prostate cancer cell adhesion to fibronectin (ECM) and filopodia formation. S3ACFL cells exhibited a significantly increased cell adhesion to fibronectin compared with control WTCFL cells (Fig. 5A). This correlated with cytoskeletal remodeling as indicated by fluorescence staining of F-actin and formation of filopodia (Fig. 5B). Confocal microscopy revealed an increased number of filopodia protrusions in S3ACFL PC-3 (arrows) compared with WTCFL PC-3 cells (Fig. 5B). High cofilin expression was detected at cell membrane regions populated by filopodia. Treatment with TGF-β led to a significant decrease in S3ACFL cell adhesion (Fig. 5A) and a reduction in filopodia protrusions (Fig. 5B). To determine whether this CFL colocalization with filopodia is dependent on endogenously derived TGF-β from the surrounding prostate CAFs (reactive stroma), we used confocal microscopy to profile the CFL/rhodamine phalloidin colocalization in S3ACFL prostate epithelial cancer cells cocultured with human CAFs. As shown in Fig. 5C, cofilin (green) colocalizes with filopodia protrusions (arrows) and loss of TGF-β resulted in increased actin/cofilin colocalization (yellow) with filopodia protrusions in S3ACFL cells in this reactive stroma–tumor microenvironment.
Figure 5.
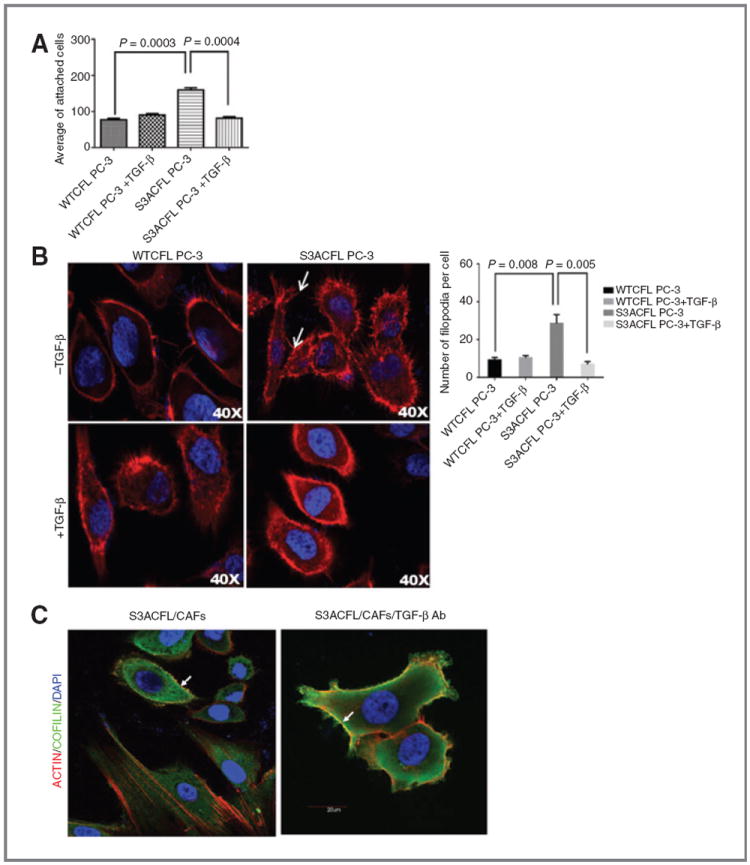
Active cofilin mediates prostate cancer cell adhesion via cytoskeletal remodeling, an effect impaired by TGF-β. A, effect of S3ACFL on prostate cancer cell adhesion. S3A mutation conferred a significant increase in cell adhesion to fibronectin compared with WTCFL cells (P = 0.0003). TGF-β significantly decreased in S3CFL cell adhesion (P = 0.0004), but not in WTCFL cells. Values shown are the mean (± SEM) of three independent experiments performed in triplicates. Statistical significance set at a P value of <0.005. B, active cofilin enhances filopodia formation; representative images of confocal microscopy (×40) show increased number of filopodia protrusions in S3ACFL PC-3 (arrows) compared with WTCFL cells. Treatment with TGF-β (5 ng/mL; 24 hours) decreased filopodia protrusions in S3ACFL cells. Filopodia quantitated in five random fields were examined for each cell line and values shown are mean ± SEM from three independent experiments (left). Statistical significance is defined at P < 0.01. C, S3ACFL active binding to F-actin at the leading edge of the cells is mediated by TGF-β. Cofilin colocalization with filopodia is dependent on TGF-β derived from surrounding stroma (CAFs). Images of cofilin/rhodamine phalloidin colocalization in S3ACFL prostate epithelial cancer cells cocultured with CAFs. Cofilin (green) colocalizes with filopodia protrusions (arrows). Loss of TGF-β (in the presence of neutralizing antibody) increases actin/cofilin colocalization (yellow) and filopodia protrusions in S3ACFL cells.
Active cofilin enhances prostate cancer metastasis in vivo
In the experimental metastasis assay, prostate cancer cells harboring the S3ACFL mutation exhibited an increased metastatic ability in vivo compared withWTCFLPC-3 prostate cancer cells, as determined by the higher number of lung metastases (Fig. 6A). Sustained exposure of prostate cancer cells with active cofilin, to TGF-β abundantly secreted in vivo, may promote their invasive properties toward metastasis (Fig. 6B).
Figure 6.
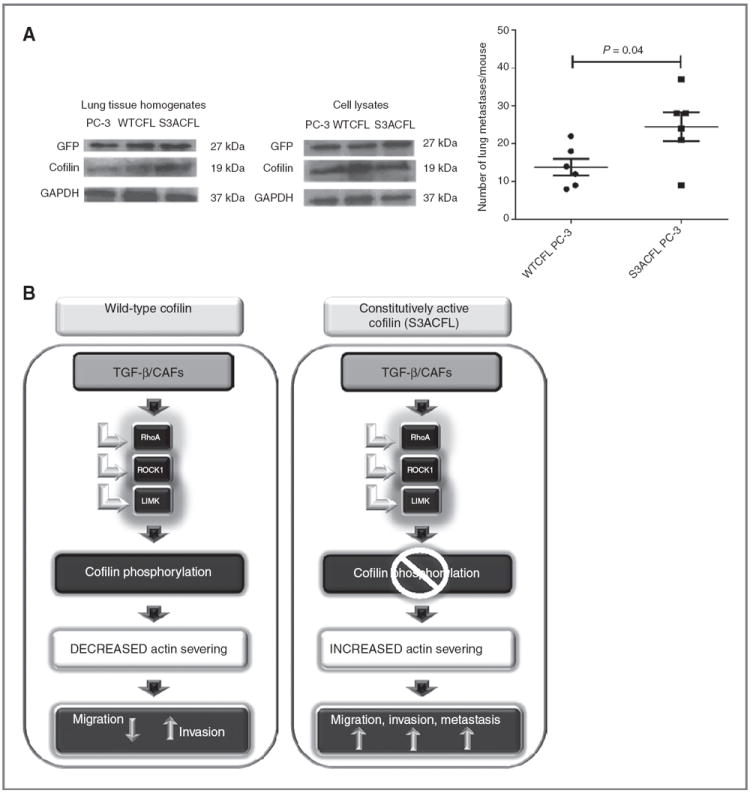
Cofilin constitutive activation promotes prostate cancer metastasis. A, male nude mice (n = 12) were inoculated with GFP-labeled PC-3 cells (parental, WTCFL, and S3ACFL) via tail vein injections. Metastatic lesions to the lungs were assessed at 4 weeks after inoculation. S3ACFL cells generated a significantly higher number of metastases compared with control PC-3 cells (P = 0.04). Values show the number of metastatic lesions to the lung/mouse for each cell line. Western blots of mouse lung tissue homogenates and cell lysates indicate the GFP presence in all samples (left). B, schematic diagram illustrating the regulatory impact of cofilin on TGF-β functional switch toward prostate cancer cell migration, invasion, and metastasis. Under constitutive activated cofilin (S3ACFL), TGF-β is produced by the reactive stroma/microenvironment (CAFs), is unable to dephosphorylate cofilin, and increases tumor cell aggressiveness and metastatic potential.
Cofilin overexpression correlates with prostate cancer progression to metastasis
TRAMP transgenic mice develop prostate adenocarcinoma with increasing age, resembling progression of human prostate cancer to metastasis. Elevated CFL expression during prostate cancer progression in the TRAMP model was associated with prostate tumor aggressiveness with increasing age (16–28 weeks; Fig. 7A). Quantitative analysis indicated a significant increase in CFL levels in metastatic tumors (28 weeks) compared with early-stage tumors and normal prostate (16 weeks WT; Fig. 7A and B). Immunohistochemical profiling of CFL in human prostate tissue specimens from a patient cohort with localized and metastatic disease to the lymph nodes and quantitative assessment of the staining (Supplementary Fig. S5) revealed a striking increase in CFL expression in metastasis compared with primary cancer in the same patient (Fig. 7C and D). Characteristic images of CFL immunoreactivity in serial sections of primary prostate tumors and metastasis are shown in Fig. 7C and D. CFL levels were significantly increased in metastasis compared with primary prostate cancer (P=0.006), whereas there were no significant differences in the expression of p-CFL or palladin proteins between primary and metastatic prostate cancer (Fig. 7E).
Figure 7.
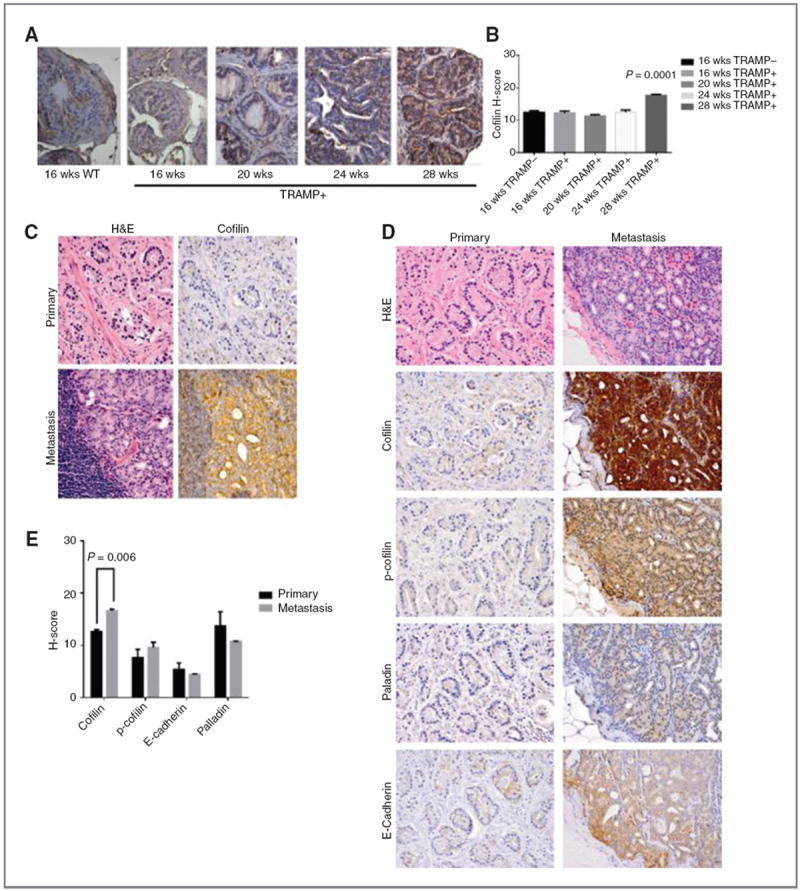
Cofilin overexpression correlates with prostate cancer progression to metastasis A and B, cofilin profiling in TRAMP mouse model. TRAMP transgenic mice develop prostate adenocarcinoma with increasing age, resembling progression of human prostate cancer to metastasis. Prostate sections of increasing grade and metastatic tumors (16–28 weeks) were profiled by immunostaining for cofilin expression; WT mouse prostate tissue (16 weeks) was used as control; magnification, ×40. Quantitative evaluation of CFL immunoreactivity, as determined by the H-scoring, shows a significant increase in metastatic tumors from 28-week-old TRAMP mice (P = 0.001) compared with early-stage tumors. C–E, cofilin expression profile in human prostate cancer. C, hematoxylin and eosin staining and CFL immunostaining in serial sections of prostate tumors; characteristic image of a metastatic lesion to lymph nodes exhibiting intense cofilin immunoreactivity, compared with the primary tumor from the same patient (absence of CFL expression). Magnification, ×100. D, representative images of immunostaining for cofilin, p-cofilin, E-cadherin, and palladin on primary and metastatic prostate cancer. E, quantitative analysis of protein immunoreactivity (from D). There was a significant increase in cofilin levels in metastatic specimens compared with primary tumors (P = 0.005).
Discussion
This study characterizes the ability of active CFL to direct prostate cancer cell migratory and invasive responses to TGF-β. The results revealed that constitutively active cofilin (S3ACFL) confers significant differences in actin remodeling proteins, migration, invasion, and metastatic potential in human prostate cancer cells in vitro and in vivo. The evidence links CFL activation to acquisition of an enhanced aggressive phenotype of prostate cancer cells in a TGF-β–dependent context. In response to TGF-β, active CFL binds to F-actin at the leading edge conferring increased tumor cell migration. Our findings that active cofilin (S3A mutation) does not exert significant consequences on prostate cancer cell invasion, but rather directs the invasive responses to TGF-β, contradictory as they might seem, are not unexpected. In breast cancer cells, CFL activation by EGF leads to an increased number of actin filament barbed ends via the polymerization of G actin monomers that generates new actin filaments and dynamic filament branching at the tip of the leading edge (43). Indeed, cell migration relies on the coordinated remodeling of the actin cytoskeleton and leading edge protrusions of moving cells are formed by lamellopodia and filopodia directing cell motility (24, 41). This study revealed a marked increase in filopodia formation in S3ACFL PC-3 cells and enhanced actin binding to the active cofilin, implicating cofilin dephosphorylation dictating prostate cancer cell migration without the regulatory effect of TGF-β, as actin severing proceeds uncontrolled. Considering that filopodia enable not only cell motility, but also facilitate attachment to the ECM and promote colonization and formation of secondary tumors at distant sites (41), our findings provide a significant insight into the ability of active CFL to promote prostate cell attachment to fibronectin by remodeling critical cell–ECM adhesion sites and metastatic dissemination in vivo. The protein–protein interactions coordinating these events are being pursued.
Our data frame a microenvironment landscape in which CFL activation (due to mutations on phosphorylation site) promotes prostate cancer cell migration and metastatic spread by increasing severing activity and actin interactions. CAFs substantially enhance the invasive properties of prostate cancer cells with constitutively active cofilin, possibly via a sustained release of TGF-β. This evidence provides a proof-ofprinciple on a direct proinvasive cross-talk between surrounding CAFs and prostate cancer cells with TGF-ß functioning as a tumor suppressor by activating the RhoA/ROCK1 signaling, leading to phosphorylation and activation of LIMK-2. This impairs cofilin severing activity, cytoskeletal reorganization, and formation of filopodia, decreasing tumor cell migration (shown schematically in Fig. 6B). During prostate cancer progression, active cofilin directs an action switching for TGF-ß to elicit metastatic responses, by enabling actin cytoskeleton remodeling and invasive tumor cell behavior. Taken together, these results provide a first insight into the role of TGF-β to impair prostate cancer growth in the early stages by putting the “breaks” on cofilin activity (via phosphorylation) and consequently compromising actin severing action. During the late stages of tumor progression, a mutation conferring constitutive activation of cofilin enables the escape to TGF-β–regulated phosphorylation by LIMK-2. In sharp contrast with an expected “action independence” from this cytokine, sustained exposure of prostate cancer cells, now harboring active cofilin (S3CFL mutation), to TGF-β secreted by the stroma microenvironment, promotes their migratory and invasive properties toward metastasis (Fig. 6B). Collectively, this evidence implies that mutational activation of cofilin occurring during prostate cancer progression is sufficient for navigating a functional switch for TGF-β from growth suppressor to a metastasis promoter. Interestingly enough, recent elegant studies implicate an additional “underestimated” contributor from the prostate microenvironment, the autonomic nerve development promoting cancer progression to metastasis (44).
The immunohistochemical profiling identified a direct association between cofilin overexpression and cancer progression to metastasis in the TRAMP mouse model of prostate tumorigenesis. More importantly of direct clinically relevance are the data demonstrating a significant increase in cofilin expression in human prostate cancer metastasis (to lymph nodes), compared with primary tumors (same patient cohort). Changes in cofilin expression have been reported in other human malignancies, including colon and ovarian cancer (45-48). Loss of the epithelial marker E-cadherin is associated with a more invasive phenotype in prostate cancer cells and high-grade and metastatic prostate cancer, as originally established (49). The strong correlation between cofilin overexpression with the in vivo metastatic and invasive properties of prostate cancer cells provides a new platform of interrogation of the cytoskeletal dynamics in cancer metastasis and the potential clinical value of cofilin in tumor progression. Indirect support for our findings is gained from recent evidence correlating cofilin with ovarian cancer progression, and a longer progression-free survival in low-expression cofilin patient cohort (47). Moreover, recent evidence supports a close functional impact of cytoskeletal dynamics on cancer metastasis under the control metabolic stress (50). Ongoing investigations are focused on two translational pursuits: (i) the correlation of cofilin and pcofilin expression with disease-free survival and biochemical recurrence in a large cohort of patients with advanced prostate cancer and (ii) the incidence of cofilin mutations (at phosphorylation sites) in human prostate tumors.
Supplementary Material
Acknowledgments
The authors thank Dr. Sergey Krupenko (Department of Biochemistry, Medical University of South Carolina, Charleston, SC) for generously providing the S3DCFL plasmid and for useful discussions; Dana Napier (Department of Pathology, University of Kentucky College of Medicine, Lexington, KY) for her excellent histologic expertise; Patrick Hensley (University of Kentucky College of Medicine) for his expertise and help in the preparation/formatting of the figures; and Lorie Howard (Division ofUrology, University of Kentucky) for her assistance in the electronic submission of the article.
Grant Support
This research was supported by the Biospecimen and Tissue Procurement Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558), an NIH grant R01 DK083761 and DOD W81XWH-08-1-0431 (N. Kyprianou), an NCI K08 grant CA155764 (C. Horbinski), 2P20 RR020171 COBRE (National Institute of General Medical Sciences), Peter and Carmen Lucia Buck Training Program in Translational Clinical Oncology (C. Horbinski), the University of Kentucky College of Medicine Physician Scientist Program (S. Larkin and C. Horbinski), and a Lyman T. Johnson Minority Fellowship (J. Collazo).
Footnotes
Authors’ Contributions
Conception and design: J. Collazo, C. Horbinski, N. Kyprianou
Development of methodology: J. Collazo, B. Zhu, C. Horbinski, N. Kyprianou
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S. Larkin, S.K. Martin, H. Pu, C. Horbinski
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J. Collazo, S. Larkin, C. Horbinski
Writing, review, and/or revision of the manuscript: J. Collazo, S. Larkin, C. Horbinski, S. Koochekpour, N. Kyprianou
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S. Koochekpour, N. Kyprianou
Study supervision: N. Kyprianou
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/)
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Arnold J, Isaacs JT. Mechanisms involved in the progression of androgen-independent prostate cancers: it is not only the cancer cell’s fault. Endocr Relat Cancer. 2002;9:61–73. doi: 10.1677/erc.0.0090061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catalona WJ. Management of cancer of the prostate. N Engl J Med. 1994;331:996–1004. doi: 10.1056/NEJM199410133311507. [DOI] [PubMed] [Google Scholar]
- 3.McKenzie S, Kyprianou N. Apoptosis evasion: the role of survival pathways in prostate cancer progression and therapeutic resistance. J Cell Biochem. 2006;97:18–32. doi: 10.1002/jcb.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karantanos T, Thompson TC. GEMMS shine a light on resistance to androgen deprivation therapy for prostate cancer. Cancer Cell. 2013;24:11–3. doi: 10.1016/j.ccr.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lunardi A, Ala U, Epping MT, Salmera L, Clohessy JG, Webster KA, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genetics. 2013;45:747–55. doi: 10.1038/ng.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuxhorn JA, Ayala GE, Smith MG, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: Induction ofmyofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res. 2002;8:2912–23. [PubMed] [Google Scholar]
- 7.Yang F, Strand DW, Rowley DR. Fibroblast growth factor-2 mediates transforming growth factor-β action in prostate cancer reactive stroma. Oncogene. 2007;27:450–9. doi: 10.1038/sj.onc.1210663. [DOI] [PubMed] [Google Scholar]
- 8.Desmoulière A, Guyot C, Gabbiani G. The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol. 2004;48:509–17. doi: 10.1387/ijdb.041802ad. [DOI] [PubMed] [Google Scholar]
- 9.Olsen CJ, Moreira J, Lukanidin EM, Ambartsumian NS. Human mammary fibroblasts stimulate invasion of breast cancer cells in a threedimensional culture and increase stroma development in mouse xenografts. BMC Cancer. 2010;10:444–60. doi: 10.1186/1471-2407-10-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin M, Soikkeli J, Jankola T, Virolainen S, Saksela O, Holtta E. TGF-β signaling, activated stromal fibroblasts and cysteine cathepsins B and L drive the invasive growth of human melanoma cells. Am J Pathol. 2011;181:2202–15. doi: 10.1016/j.ajpath.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 11.Nasu Y, Timme TL, Yang G, Bangma CH, Li L, Ren C, et al. Suppression of caveolin expression induces androgen sensitivity in metastatic androgen-insensitive mouse prostate cancer cells. Nat Med. 1998;4:1062–4. doi: 10.1038/2048. [DOI] [PubMed] [Google Scholar]
- 12.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 1996;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 13.Goel H, Li J, Kogan S, Languino LR. Integrins in prostate cancer progression. Endocr Relat Cancer. 2008;15:657–64. doi: 10.1677/ERC-08-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010;16:1414–20. doi: 10.1038/nm.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. J Mol Aspects Med. 2010;31:205–14. doi: 10.1016/j.mam.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar J, Park S-H, Cieply B, Shupp J, Killiam E. A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol Cell Biol. 2011;31:4036–51. doi: 10.1128/MCB.01342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Condeelis JS, Singer RH, Segall J. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718. doi: 10.1146/annurev.cellbio.21.122303.120306. [DOI] [PubMed] [Google Scholar]
- 18.Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Davila M, Frost AR, Grizzle WE, Chakrabarti R. LIM kinase 1 is essential for the invasive growth of prostate epithelial cells: implications in prostate cancer. J Biol Chem. 2003;278:36868–75. doi: 10.1074/jbc.M306196200. [DOI] [PubMed] [Google Scholar]
- 20.Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- 21.Lee SH, Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Mol Cell. 2010;29:311–25. doi: 10.1007/s10059-010-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 2010;8:629–42. doi: 10.1158/1541-7786.MCR-10-0139. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Godt D, Gunsalus K, Kiss I, Goldberg M, Laski FA. Cofilin/ADF is required for cell motility during Drosophila ovary development and oogenesis. Nat Cell Biol. 2001;3:204–9. doi: 10.1038/35055120. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–6. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- 25.DesMarais V, Sidani M, Scemes E, Wang W, Song X, Eddy R, et al. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr Biol. 2006;16:2193–205. doi: 10.1016/j.cub.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008;87:649–67. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Hotulainen P, Paunola E, Vartiainen MK, Lappalainen P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell. 2005;16:649–64. doi: 10.1091/mbc.E04-07-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu Rev Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- 29.Olson MF. Applications for ROCK kinase inhibition. Curr Opin Cell Biol. 2008;20:242–8. doi: 10.1016/j.ceb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oleinik NV, Krupenko NI, Krupenko SA. ALDH1L1 inhibits cell motility via dephosphorylation of cofilin by PP1 and PP2A. Oncogene. 2010;25:6233–44. doi: 10.1038/onc.2010.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jakowlew SB. Transforming growth factor-β in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–57. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 32.Teicher BA. Transforming growth factor-beta and the immune response to malignant disease. Clin Cancer Res. 2007;13:6247–51. doi: 10.1158/1078-0432.CCR-07-1654. [DOI] [PubMed] [Google Scholar]
- 33.Pu H, Collazo J, Jones E, Gayheart D, Sakamoto S, Vogt A, et al. Dysfunctional TGF-β receptor II accelerates prostate tumorigenesis in the TRAMP mouse model. Cancer Res. 2009;69:7366–74. doi: 10.1158/0008-5472.CAN-09-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo Y, Kyprianou N. Overexpression of transforming growth factor (TGF)-β1 type II receptor, restores TGF-β1-sensitivity and signaling in human prostate cancer cells, LNCaP. Cell Growth and Differentiation. 1998;9:185–93. [PubMed] [Google Scholar]
- 35.Bello-Deocampo D, Tindall DJ. TGF-β/Smad signaling in prostate cancer. Curr Drug Targets. 2003;4:197–207. doi: 10.2174/1389450033491118. [DOI] [PubMed] [Google Scholar]
- 36.Massagué J. TGF β in Cancer. Cell. 2008;25:215–30. [Google Scholar]
- 37.Micalizzi DS, Wang CA, Farabaugh SM, Schiemann WP, Ford HL. Homeoprotein Six1 increases TGF-beta type I receptor and converts TGF-beta signaling from suppressive to supportive for tumor growth. Cancer Res. 2010;1570:10371–80. doi: 10.1158/0008-5472.CAN-10-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu B, Fukada K, Zhu H, Kyprianou N. Prohibitin and cofilin are intracellular effectors of transforming growth factor-β signaling in human prostate cancer cells. Cancer Res. 2006;66:8640–77. doi: 10.1158/0008-5472.CAN-06-1443. [DOI] [PubMed] [Google Scholar]
- 39.Yang F, Strand DW, Rowley DR. Fibroblast growth factor-2 mediates transforming growth factor-β action in prostate cancer reactive stroma. Oncogene. 2007;27:450–9. doi: 10.1038/sj.onc.1210663. [DOI] [PubMed] [Google Scholar]
- 40.Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumors promotes metastasis. Nat Commun. 2013;4:1795–805. doi: 10.1038/ncomms2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arjonen A, Kaukonen R, Ivaska J. Filopodia and adhesion in cancer cell motility. Cell Adh Migr. 2011;5:421–30. doi: 10.4161/cam.5.5.17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu H, Zhao J, Zhu B, Collazo J, Gal J, Shi P, et al. EMMPRIN regulates cytoskeleton reorganization and prostate cancer cell invasion. Prostate. 2012;72:72–81. doi: 10.1002/pros.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zebda N, Bernard O, Bailly M, Welti S, Lawrence DS, Condeelis JS. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipodia extension. J Cell Biol. 2000;27:1119–28. doi: 10.1083/jcb.151.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer metastasis. Science. 2013;341:361–7. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 45.Wang W, Mouneimne G, Sidani M, Wyckoff J, Chen X, Makris A, et al. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol. 2006;8:395–404. doi: 10.1083/jcb.200510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W, Eddy R, Condeelis JS. The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer. 2007;7:429–40. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishimura S, Tsuda H, Kataoka F, Arao T, Nomura H, Chiyoda T, et al. Overexpression of cofilin 1 can predict progression-free survival in patients with epithelial ovarian cancer receiving standard therapy. Human Pathol. 2011;42:516–21. doi: 10.1016/j.humpath.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 48.Popow-Woźniak A, Mazur AJ, Mannherz HG, Malicka-Bøaszkiewicz M, Nowak D. Cofilin overexpression affects actin cytoskeleton organization and migration of human colon adenocarcinoma cells. Histochem Cell Biol. 2012;138:725–36. doi: 10.1007/s00418-012-0988-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Umbas R, Schalken JA, Aalders TW, Carter BS, Karthaus HF, Schaafsma HE, et al. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate. Cancer Res. 1992;52:5104–10. [PubMed] [Google Scholar]
- 50.Caino CM, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest. 2013;123:2907–20. doi: 10.1172/JCI67841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.