

Abstract
Black pepper is one of the most popular and oldest spices in the world and valued for its pungent constituent alkaloids. Pinerine is the main bioactive compound in pepper alkaloids, which perform unique physiological functions. However, the mechanisms of piperine synthesis are poorly understood. This study is the first to describe the fruit transcriptome of black pepper by sequencing on Illumina HiSeq 2000 platform. A total of 56,281,710 raw reads were obtained and assembled. From these raw reads, 44,061 unigenes with an average length of 1,345 nt were generated. During functional annotation, 40,537 unigenes were annotated in Gene Ontology categories, Kyoto Encyclopedia of Genes and Genomes pathways, Swiss-Prot database, and Nucleotide Collection (NR/NT) database. In addition, 8,196 simple sequence repeats (SSRs) were detected. In a detailed analysis of the transcriptome, housekeeping genes for quantitative polymerase chain reaction internal control, polymorphic SSRs, and lysine/ornithine metabolism-related genes were identified. These results validated the availability of our database. Our study could provide useful data for further research on piperine synthesis in black pepper.
Introduction
Black pepper, which is known as the “King of Spices”, is one of the most important and popular spices in the world. Black pepper is an important member of the Piperaceae family originating from the southwestern region of India, where its trade was initially restricted [1]. Black pepper, with its characteristic pungency and flavor, has been used as an ingredient in many food preparations for thousands of years. As one of the most extensively used spice in the world, black pepper is cultivated in tropical areas. The volume of black pepper trade reached 1.9 billion US dollars from a production of 4.6 × 105 tons in 2012 (Food and Agriculture Organization data). Black pepper is used not only in human diet but also in other applications, such as medicine, preservative, perfumery, and insecticide, because this species exhibits antioxidant, anti-inflammatory, and anticancer properties [2–4].
The significant value of black pepper is due to the presence of piperidine alkaloids. Piperine (1-piperoylpiperidine) is a pungent, nitrogenous substance and major alkaloid in black pepper [5]. Piperine, which is utilized as a gustatory enhancer, is an effective agonist at the transient receptor potential cation channel subfamily V member 1 receptor, evoking a painful burning sensation sufficient to be a deterrent to most animals [6,7]. By contrast, dietary intake of piperine significantly stimulates the digestive enzymes of the pancreas and the intestines. As a result, digestive capacity is enhanced and gastrointestinal food transit time is shortened [8,9]. With unique taste and digestive function of piperine, black pepper has been considered as a popular food seasoning worldwide. In medical research, piperine has been validated in in vitro experiments to protect against oxidative damage by quenching reactive oxygen species and inhibiting lipid peroxidation [10,11]. Despite dietary and medical importance of piperine in black pepper, the underlying molecular mechanism remains unclear. Previous research showed that piperine is derived from the products of the primary metabolism of lysine/ornithine, which has been extensively studied in model plants [12,13]. However, further research on piperine biosynthesis mechanism in black pepper as a non-model plant is limited because of lack of an available molecular database.
As a non-model plant, black pepper does not have available genomic information. In the absence of a sequenced genome, de novo assembly of RNA-Seq is a cost-effective method to study the transcriptomes of most organisms [14,15]. Millions of short tags have been generated from RAN-Seq platform, such as Roche 454, Illumina Genome Analyzer, and Applied Biosystems SOLiD. After short tags are assembled using specific tools and algorithms, genome and transcriptome sequences are interpreted. Sequence platform, assembly tools, and special bioinformatics analysis can be applied to generate a complex technology system for RNA-Seq, which can identify transcript sequence polymorphisms, novel trans-splicing, and splice isoforms [16–18]. Compared with traditional approaches, RNA-Seq technologies provide highly specific and quantitative measurements.
In this study, a fruit transcriptome of black pepper was analyzed using Illumina HiSeq 2000 platform. Approximately 56 million clean reads were generated and 44,061 de novo assembled unigenes were obtained. The housekeeping gene, lysine/ornithine-related genes, and some polymorphic simple sequence repeat (SSR) primers were identified. These results, which constitute the first dataset of the sequence of the black pepper fruit, provide a useful gene library for black pepper molecular research.
Materials and Methods
Plant materials and nucleic acid isolation
Plant materials were obtained from 10 year-old black pepper cultivar (Piper nigrum L. cv. ‘Reyin No. 1’) with collecting permit in Spice and Beverage Research Institute of Chinese Academy of Tropical Agricultural Science, Wanning, Hainan, China. Eight fruits of black pepper were collected every month after pollination. Total RNA was isolated individually using TRIzol reagent (Life Technologies). The high quality RNA were obtained through twice Chloroform/Isoamyl alcohol (24:1) purification, and then mixed with approximately the same quantity. Total 5μg mRNA was purified using poly-T oligo-attached magnetic beads (Life Technologies) for RNA-Seq. Different tissues of black pepper were collected (root, stem, leaf, flower, and fruits). Total RNA was also isolated from each tissue for further expression analysis.
Illumina cDNA library construction and sequencing
An Illumina HiSeq 2000 library was constructed for Solexa sequencing. The enriched poly(A) mRNA was fragmented into small pieces of 200 bp to 700 bp by using divalent cations at 75°C. First-strand cDNA was synthesized by reverse transcriptase with random hexamer primers. Second-strand cDNA was subsequently synthesized by DNA Polymerase I and RNase H (Invitrogen). The cDNA fragments were ligated to sequencing adapters and analyzed through agarose gel electrophoresis to select suitable fragments for enrichment by polymerase chain reaction (PCR) amplification. The resulting cDNA library was sequenced on Illumina HiSeq 2000 platform in Beijing Genomics Institute Genomic Center in Shenzhen, China (http://www.genomics.cn).
Sequence assembly and functional annotation
Total raw reads from sequencing were preprocessed to remove dirty raw reads, including (1) adapters that were added for reverse transcription and sequencing, (2) sequences with unknown nucleotides larger than 5%, and (3) low-quality reads (the rate of reads with quality value of ≤10 is more than 20%). The filtered clean reads were then assembled using Trinity method (http://trinityrnaseq.sourceforge.net/) [14]. Overlapping information in the short reads was used to construct contigs with high coverage. Afterward, the reads were mapped back to the contigs to connect these contigs and to identify the sequences that cannot be extended on either end. Such sequences were defined as unigenes. Optimal results were selected on the basis of evaluation results of the assembly encompassing the total number of unigenes, the distribution of unigene length, the N50 statistic, and the average coverage.
We conducted a BLAST search against the non-redundant protein (NR) and nucleotide sequences (NT) databases in the National Center for Biotechnology Information (NCBI), Swiss-Prot, and Clusters of Orthologous Groups (COG) with an E value cutoff of 10−5 to assign putative functions to the unigenes. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) classifications were conducted by using Blast2GO according to Gotz et al. [19]. Gene names were assigned to each unigene based on the best hit (highest score).
Internal control gene identification
The housekeeping genes were identified from transcriptome data. The housekeeping genes were searched through gene descriptions in functional annotation based on the candidate gene name. The first-strand cDNA template of different tissues (root, stem, leafy, flower, and fruits) was synthesized using a reverse transcriptase kit (Thermo) in accordance with the manufacturer’s instructions. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed using ABI 7500 Real-time PCR system (Applied Biosystems, Foster City, CA, USA) in accordance with the manufacturer’s instructions. For each analysis, qRT-PCR assays were conducted in three sample replications. Error bars denoted the standard error of three replications.
For internal control gene identification, Microsoft Excel file of raw expression values for the tested genes in different samples was imported into geNORM to analyze gene expression stability [20]. The specificity of amplifications was verified by melting curve analysis (60°C to 95°C) after 40 cycles.
SSR detection
SSR was detected with MIcroSAtellite software in which unigenes were used as reference data. SSRs with length of more than 150 bp on both ends of the unigene were retained, and these sequences were used to design primers in Primer Premier 6.0 (PREMIER Biosoft International, Palo Alto, CA, USA). The primers were filtered by removing the following: (1) primers without SSRs; (2) primers aligned to unigene sequences with more than three mismatches in the 5′ site and one mismatch in the 3′ site; (3) primers aligned to more than one unigene.
Twelve Piperaceae species were used in polymorphism analysis (S4 Table). DNA was isolated according to the cetyl trimethylammonium bromide protocol (CTAB). PCR was conducted to determine the polymorphism of primers. The following thermal cycling conditions were applied: 95°C for 5 min and 35 cycles of 95°C for 60 s, 55°C for 30 s, and 72°C for 90 s. The PCR product was analyzed by denaturing polyacrylamide gel electrophoresis.
Results
Sequencing and de novo transcriptome assembly
The fruits of black pepper (P. nigrum L. cv. Reyin No. 1) at different developmental stages were collected at 1 month to 10 months post anthesis. The RNA of these fruits was isolated. RNAs of equal quality were mixed for Illumina sequencing. A total of 56,281,710 raw reads were obtained. We filtered the sequence data for clean reads, resulting in 52,098,738 clean reads. All clean reads were de novo assembled into contigs by using the Trinity method [14]. The clean read assembly generated 179,075 contigs when all isoforms were included. These contigs represent a total of 44,061 unigenes that were considered for downstream analysis with an N50 length of 1,757 nt. The length of the unigenes ranged from 300 nt to 15,000 nt, with a mean length of 1,354 nt and 46.60% GC content (Table 1). A total of 23,085 (52.39%) unigenes longer than 1 kb and 9,769 (22.17%) unigenes longer than 2 kb were obtained. The length distributions of the unigenes are shown in Table 1; the results revealed that more than 30,000 unigenes (75.77%) were longer than 500 nt (Table 1). The database was deposited in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/) under accession number SRS856941.
Table 1. Summary of transcriptome data for black pepper fruits.
| Number | Percentage | |
|---|---|---|
| Raw reads | 56,281,710 | |
| Clean reads | 52,098,738 | |
| Contigs | 179,075 | |
| Total unigenes | 44,061 | |
| Total sizes (nt) | 59,262,045 | |
| Unigenes (300–500 nt) | 10,678 | 24.23% |
| Unigenes (500–1000 nt) | 10,298 | 23.38% |
| Unigenes (1000–2000 nt) | 13,316 | 30.22% |
| Unigenes (2000–5000 nt) | 9,409 | 21.35% |
| Unigenes (>5000 nt) | 360 | 0.82% |
| Mean length (nt) | 1,345 | |
| GC content N50 length | 1,757 | 46.60% |
| Unigenes in NR database | 32,697 | 74.20% |
| Unigenes in NT database | 25,366 | 57.57 |
| Unigenes in SwissProt database | 23,080 | 52.38% |
| Unigenes in GO database | 28,827 | 65.43% |
| Unigenes in COG database | 16,195 | 36.76% |
| Unigenes in KEGG database | 24,836 | 56.37% |
Gene annotation and functional classification
The homology-based approach was adopted in functional annotation. All unigenes were searched against the NCBI NR and NT databases through BLASTX searches in which an E value cutoff of 10−5 was set. A total of 32,697 (74.20%) unigenes showed significant similarity to known proteins in the NR database. Likewise, a total of 25,366 (57.57%) exhibited significant similarity to known nucleotide sequences in the NT database. Sequences were also searched against the Swiss-Prot database to detect additional reference annotations, resulting in 23,080 (52.38%) annotated unigenes (Table 1). In the analysis of the matching sequences based on NR annotation, E value distribution showed that 55.80% of the annotated sequences displayed strong homology (E value less than 1E − 45; Fig 1A). In species distribution analysis, Vitis vinifera was ranked first with 12,948 (39.68%) top BLAST hits, followed by Ricinus communis, Prunus persica, Populus trichocarpa, and Glycine max with 2,813 (8.62%), 2,809 (8.60%), 2,332 (7.15%), and 1,632 (5%) top BLAST hits, respectively (Fig 1B).
Fig 1. Summary results of NR sequence annotation.
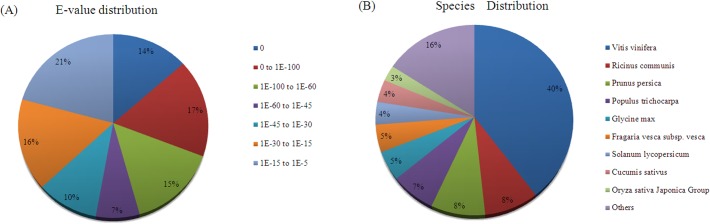
(A) E value distribution of BLASTX matches for each unigene. (B) Species-based distribution of BLASTX matches for each unigene.
As the international standardized gene functional classification system, GO and COG classifications were conducted for transcriptome data annotation. A total of 28,827 (65.43%) unigenes were assigned to 54 Level 2 GO terms, which were summarized under three main GO categories, including biological process, cellular component, and molecular function (Fig 2). Among all of the categories, cellular processes and metabolic processes in the biological processes, cell and cell part in the cellular component, and binding and catalytic activity in the molecular function represented the major subcategories. In the COG classification, 16,195 unigenes were classified into 24 COG categories. At the top, the clusters included general function prediction only, replication, and transcription (Fig 3).
Fig 2. Distribution of GO classification (Level 2).

Annotated unigenes were classified into 3 major categories (biological processes, cellular components, and molecular function) and 54 subgroups. The x-axis indicates the subgroups in GO annotation. The y-axis indicates the percentage of specific categories of genes in each main category.
Fig 3. Distribution of COG classification.

The unigenes with significant homologies in the COG database were classified into 25 COG categories. The x-axis indicates the subgroups in the COG classification. The y-axis indicates the number of genes in each main category.
All of these functional annotation assignments provide valuable information to investigate specific biochemical and developmental processes in fruit development of black pepper. The entire annotation information of transcriptome data is shown in S1 Table.
KEGG pathway enrichment and piperine-related gene scanning
KEGG is a pathway-based categorization of orthologous genes that provide information to predict the functional profiles of genes. In this study, 24,836 genes were mapped into 128 signaling pathways to further validate the biological pathways that are active in the black pepper fruit. Among these genes, 6,558 (26.4%) unigenes were involved in the metabolic pathway, which was the main group in pathway enrichment, followed by biosynthesis of secondary metabolites (2,399; 9.7%), endocytosis (1,772; 7.1%), and glycerophospholipid (1,711; 6.9%) (S2 Table). These results provided a valuable resource that could be used to investigate specific processes and pathways in the development of black pepper fruit.
A previous study showed that piperidine alkaloids are derived from the products of lysine/ornithine metabolism [21]. The unigenes with full functional domain, annotated in the lysine/ornithine metabolism-related pathway, were identified manually. A total of 17 typical genes were identified as potentially related to lysine/ornithine metabolism, including lysine/ornithine decarboxylase, lysine dehydrogenase, and primary amine oxidase (Table 2). These genes might participate in piperidine, quinolizidine, indolizidine, and lycopodium alkaloid biosynthesis, which will provide valuable resource for further research.
Table 2. The list of lysine mechanism-related genes in transcriptome.
| Gene ID | Length | KO_id | ko_definition |
|---|---|---|---|
| CL3230.Contig3_F0463-PN | 1711 | K01581 | ornithine decarboxylase [EC:4.1.1.17] |
| CL1221.Contig2_F0463-PN | 1806 | K01586 | diaminopimelate decarboxylase [EC:4.1.1.20] |
| CL16778.Contig2_F0463-PN | 1610 | K01581 | ornithine decarboxylase [EC:4.1.1.17] |
| CL44.Contig2_F0463-PN | 2878 | K00276 | primary-amine oxidase [EC:1.4.3.21] |
| CL9334.Contig1_F0463-PN | 2082 | K13367 | non-specific polyamine oxidase [EC:1.5.3.17] |
| Unigene14041_F0463-PN | 2504 | K00276 | primary-amine oxidase [EC:1.4.3.21] |
| CL13005.Contig1_F0463-PN | 1978 | K00818 | acetylornithine aminotransferase [EC:2.6.1.11] |
| Unigene14591_F0463-PN | 2315 | K00276 | primary-amine oxidase [EC:1.4.3.21] |
| Unigene7223_F0463-PN | 2206 | K00276 | primary-amine oxidase [EC:1.4.3.21] |
| CL9890.Contig2_F0463-PN | 1293 | K01778 | diaminopimelate epimerase [EC:5.1.1.7] |
| CL3253.Contig4_F0463-PN | 1552 | K13065 | shikimate O-hydroxycinnamoyltransferase [EC:2.3.1.133] |
| CL3823.Contig1_F0463-PN | 1696 | K01438 | acetylornithine deacetylase [EC:3.5.1.16] |
| CL3485.Contig1_F0463-PN | 2233 | K00928 | aspartate kinase [EC:2.7.2.4] |
| CL13479.Contig1_F0463-PN | 6480 | K11446 | histone lysine-demethylase JARID1 [EC:1.14.11.-] |
| CL5146.Contig2_F0463-PN | 3370 | K15601 | lysine-specific demethylase 3 [EC:1.14.11.-] |
| CL14382.Contig2_F0463-PN | 1349 | K00215 | dihydrodipicolinate reductase [EC:1.3.1.26] |
| CL1221.Contig2_F0463-PN | 1806 | K01586 | diaminopimelate decarboxylase [EC:4.1.1.20] |
Internal control gene identification
Internal control genes were utilized for further analysis to assess transcriptome quality and provide fundamental basis for specifically expressed gene scanning. In this study, seven frequently used internal control genes were selected from the transcriptome annotation data by using key word search. These internal control genes were histone H3, ubiquitin-7, cyclophilin, polyubiquitin-1, polyubiquitin-2, glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH), and actin. The primers were designed with amplification lengths of 100 nt to 200 nt and melting temperatures (T m) of 55°C to 60°C (Table 3). The expression stabilities of these seven genes were assessed by two-step real-time PCR in a set of five different tissue samples. The dissociation curves showed one specific peak, which indicated the specific amplification of PCR (Fig 4A). The cycle threshold (C t) values of these seven internal control genes in different tissues were also obtained through real-time PCR amplification, which varied not so obviously (Fig 4B). Then, geNORM was applied to identify the best internal control genes in various tissue samples. geNORM is a statistical algorithm that determines gene stability measure (M) of all of the genes under investigation based on the C t value in different samples; a lower M value corresponds to a more stable expression [20]. After C t value was evaluated, the results showed that the average expression M value of histone H3 was the lowest and most suitable internal control gene for black pepper (Fig 4C). Therefore, histone H3 can be selected as a reference gene for tissue-specific gene analysis.
Table 3. The primers of internal control genes for real-time PCR analysis.
| Gene name | Gene NO. | Primers (5`-3`) |
|---|---|---|
| Histone 3 | CL15850.Contig2_F0463-PN | GAAGTCAGCCCCGACGACAGGT |
| CGAACGAGACGCTGGAAAGGTA | ||
| Ubiquitin 7 | Unigene17089_F0463-PN | CCCCAGACCAGCAGCGTTTAATC |
| CATCGACCTTGTAGAACTGCAGGAC | ||
| Actin | CL6615.Contig1_F0463-PN | GAAACTGGGTATCTGTGAGGCTGA |
| GCAAGTGCTTCCTGATGAACAACA | ||
| GAPDH | CL9041.Contig3_F0463-PN | CTTTCTGTAGCCAACTCCTCTCTCC |
| GACGGAGAAGAAGTCATCGGAAG | ||
| Cyclophilin | Unigene5924_F0463-PN | CCATTTGTGTCAGACCCAGCATT |
| GGTGATGGTAGAGGAGGGGAGTC | ||
| Polyubiquitin-1 | Unigene8228_F0463-PN | TTACCAGGACTCAGCAGCGAATG |
| AAGCCAATGACTTTACATCCTCCAG | ||
| Polyubiquitin-2 | CL13523.Contig3_F0463-PN | AGGAACGAGTTGAAGAGAAAGAAGG |
| TCCACCCCGTAGAGCCAGAACAAG |
Fig 4. Identification of internal control genes.

(A) Dissociation curves of seven genes in different samples. One specific peak represents the specific amplification of genes. (B) C t values of genes in different samples. (C) Average expression stability values of genes, in which the lowest mean M value of histone H3 indicates the most stable expression.
Detection of SSR markers
SSR served as the most important molecular marker, which has been extensively utilized for gene mapping, molecular breeding, genetic diversity, and discrimination. A total of 5,509 SSRs loci with dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeats were detected in 5,252 unigenes by scanning the transcriptome data. Among these SSRs, the trinucleotide repeat motifs were the most abundant, accounting for 3,557 SSRs (64.57%), followed by 1,607 (29.17%) dinucleotide repeat motifs, 130 (2.36%) hexanucleotide repeats, 116 (2.10%) pentanucleotide repeat motifs, and 99 (1.80%) tetranucleotide repeat motifs (Table 4). The main motifs were the dinucleotide AG/CT repeat (681) and AT/TA repeat (596) and the trinucleotide CCG/CGG repeat (1,058) and AGG/CCT repeat (545). Detailed information of the SSR type is shown in Fig 5.
Table 4. Summary of SSR searching results.
| Searching Item | NO. |
|---|---|
| Total number of unigenes examined | 44,061 |
| Total size of examined sequences (nt) | 59,262,045 |
| Total number of identified SSRs | 5,509 |
| Number of SSR containing unigenes | 5,252 |
| Number of sequences containing more than 1 SSR | 217 |
| Di-nucleotide | 1,607 |
| Tri-nucleotide | 3,557 |
| Tetra-nucleotide | 99 |
| Penta-nucleotide | 116 |
| Hexa-nucleotide | 130 |
Fig 5. Summary of the SSR types in the transcriptome.

A total of 5,509 SSRs were identified. The x-axis indicates the repeat type. The y-axis indicates the number of different repeats.
Based on the SSR-containing unigenes, the SSR primers were designed using Primer Premier 6.0 (PREMIER Biosoft International, Palo Alto, CA, USA) according to Wang et al. [22]. In total, 3,681 pairs of primers were obtained (S3 Table). A total of 24 pairs of primers were randomly selected for PCR amplification, and 12 Piperaceae species were selected for polymorphism analysis of the primers. The results showed that 11 pairs of primers exhibited ideal polymorphisms (S4 Table). These SSR primers could represent a valuable biomarker resource of P. nigrum. However, all putative SSR primers should be validated before use.
Discussion
As one of the most popular and oldest spices, black pepper has been used in diet, perfumery, and medicine for thousands of years [23]. However, molecular biology research and available gene data for molecular research are limited [24–27]. Nevertheless, high-throughput RNA-Seq is an effective method to obtain large amounts of transcriptome data from different tissue types. In this study, the transcriptome of black pepper fruit was described for the first time. High-quality transcriptome data were obtained using Illumina HiSeq 2000 sequencing platform and assembled using the Trinity method with multiple optimal k-mer lengths and cutoff values. The assembly strategy was considered as a unified solution for transcriptome construction in any sample, particularly in the absence of a reference genome [14,28,29]. In sequence assembly, the N50 length was used to evaluate assemblies in which a high number corresponds to high quality. The high quality of our data was confirmed by high value of N50 and average length in sequence assembly (N50 = 1,575 bp, average length = 1,345 bp); this result was comparable to that obtained in published transcriptomic analyses of other plant species, such as Reaumuria soongorica (N50 = 1,109 bp, average length = 677 bp), German cockroach (N50 = 792 bp, average length = 798 bp), and Haloxylon ammodendron (N50 = 1,345 bp, average length = 728 bp) [30–32].
Functional annotation and classification were introduced to illustrate the transcriptome comprehensively. A large number of unigenes were annotated with molecular function by searching against the NCBI NR and NT and Swiss-Prot databases. Moreover, the annotated genes of black pepper showed higher homology to V. vinifera (Fig 1). The species distribution in annotation might reveal the evolutionary relationship of black pepper with other species. With specific metabolic alkaloids, black pepper has been considered as a common species among spices [10]. GO and COG classifications provided further insights into the role of metabolic alkaloids in the development of black pepper fruit. The main subcategories of gene distribution in cellular component and molecular function of GO annotation were similar to those of other species. In biological process, the high proportion of metabolic and cellular processes indicated the unique characteristic of black pepper development (Fig 2) [32,33].
Although RNA-Seq technology is an efficient method to describe the transcriptomes, further experiments were required to verify the utility of data. In this study, we focused on internal control gene identification in transcriptome data. An appropriate internal control should be determined to standardize the quantitative expression of different genes. Housekeeping genes, such as actin, tubulin, histone, and GAPDH, were most frequently applied for their stable expression in different tissues and conditions [34–36]. However, the transcript levels of these genes are not always stable. The suitable internal control genes for new species must be determined before use. Statistical algorithms, such as geNORM or BestKeeper, have been developed to determine the best suitable genes [20,37]. Housekeeping genes in our transcriptome data were searched by gene annotation. Seven sequences with correct annotation and full-length protein domain were selected for subsequent study (Table 3). The specific amplification and expression data were obtained through real-time PCR; histone H3 (CL15850.Contig2_F0463-PN) was defined as the most suitable internal control gene of black pepper (Fig 4). In addition, large amounts of SSR markers and primers were identified from the transcriptome data (S3 Table). The polymorphisms of 24 pairs of primers were randomly determined by denaturing polyacrylamide gel. Approximately half of the polymorphisms could be used in related studies. To our knowledge, the polymorphic SSR markers are important molecular tools, which have been extensively utilized for genetic diversity, gene mapping, molecular breeding, and gene-based association studies [38]. Our study on internal control gene and polymorphic SSR marker identification not only illustrates the usefulness of our transcriptome data but also provides available reference data for further research.
Black pepper is well known for its specific alkaloids in fruits. The most important functional substance among piperidine alkaloids was piperine, which is accounted for the pungent taste and the medicinal effect of black pepper [25,39]. We described the transcriptome of black pepper fruit to investigate piperine biosynthesis. As a lysine-derived alkaloid, piperine undergoes several steps, including lysine decarboxylation as the first step in alkaloid biosynthesis in which cadaverine is produced [40]. Cadaverine is then used as a functional substance to synthesize piperidine through a series of reactions, including oxidation, dehydration, and cyclization [41–43]. A previous study showed that piperidine is the direct precursor of piperine synthesis [12]. Although we determined the main line of the piperine synthetic pathway from different research results, a systemic study on piperine synthesis and key gene identification is limited. In KEGG enrichment, potential genes involved in lysine metabolism were initially selected. Based on the overall consideration of annotation and sequence information, the unigenes showed consistent functional description. The full-length protein domain is shown in Table 2. The genes presented in this study would provide putative targets for further research on piperine synthesis.
Conclusion
This study presents the first transcriptome sequencing analysis of black pepper fruit using Illumina RNA-Seq technology. A total of 44,061 unigenes with an average length of 1,345 nt were generated and 40,537 were annotated. Based on these annotated unigenes, the characteristic of the transcriptome was illustrated comprehensively by bioinformatics analysis. The subsequent work of lysine metabolism related gene, internal control gene and polymorphic SSR identification suggest the availability of transcriptome data. Our study highlight the potential of RNA-seq for functional genomics researches on different species which genomic sequence data are not available. All of these data provide fundamental reference for further functional genomics studies on black pepper.
Supporting Information
(XLSX)
(XLS)
(XLSX)
(DOC)
Acknowledgments
Financial support from the Ministry of Agriculture of the People’s Republic of China(2015NWB046, 15RZZYS-13) are appreciated.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This project was funded by the Ministry of Agriculture of the People’s Republic of China (2015NWB046, 15RZZYS-13). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nair KP (2011) Agronomy and Economy of Black Pepper and Cardamom The King and Queen of Spices: Elsevier press. 380 p. [Google Scholar]
- 2. Gülçin İ (2005) The antioxidant and radical scavenging activities of black pepper (Piper nigrum) seeds. International journal of food sciences and nutrition 56: 491–499. [DOI] [PubMed] [Google Scholar]
- 3. Bothi Raja P, Sethuraman MG (2008) Inhibitive effect of black pepper extract on the sulphuric acid corrosion of mild steel. Materials letters 62: 2977–2979. [Google Scholar]
- 4. Vijayan K, Thampuran RA (2000) Pharmacology, toxicology and clinical application of black pepper Black pepper: CRC Press; pp. 455–466. [Google Scholar]
- 5. Srinivasan K (2007) Black pepper and its pungent principle-piperine: a review of diverse physiological effects. Critical reviews in food science and nutrition 47: 735–748. [DOI] [PubMed] [Google Scholar]
- 6. McNamara FN, Randall A, Gunthorpe MJ (2005) Effects of piperine, the pungent component of black pepper, at the human vanilloid receptor (TRPV1). British journal of pharmacology 144: 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okumura Y, Narukawa M, Iwasaki Y, Ishikawa A, Matsuda H, Yoshikawa M, et al. (2009) Activation of TRPV1 and TRPA1 by black pepper components. Bioscience, biotechnology, and biochemistry 74: 1068–1072. [DOI] [PubMed] [Google Scholar]
- 8. Mehmood MH, Gilani AH (2010) Pharmacological basis for the medicinal use of black pepper and piperine in gastrointestinal disorders. Journal of medicinal food 13: 1086–1096. 10.1089/jmf.2010.1065 [DOI] [PubMed] [Google Scholar]
- 9. Platel K, Srinivasan K (2000) Influence of dietary spices and their active principles on pancreatic digestive enzymes in albino rats. Food/Nahrung 44: 42–46. [DOI] [PubMed] [Google Scholar]
- 10. Zarai Z, Boujelbene E, Ben Salem N, Gargouri Y, Sayari A (2013) Antioxidant and antimicrobial activities of various solvent extracts, piperine and piperic acid from Piper nigrum . LWT-Food Science and Technology 50: 634–641. [Google Scholar]
- 11. Prasad NS, Raghavendra R, Lokesh BR, Naidu KA (2004) Spice phenolics inhibit human PMNL 5-lipoxygenase. Prostaglandins, Leukotrienes and Essential Fatty Acids 70: 521–528. [DOI] [PubMed] [Google Scholar]
- 12. Geisler JG, Gross GG (1990) The biosynthesis of piperine in Piper nigrum. Phytochemistry 29: 489–492. [Google Scholar]
- 13. Szőke É, Lemberkovics É, Kursinszki L (2013) Alkaloids Derived from Lysine: Piperidine Alkaloids Natural Products: Springer press; pp. 303–341. [Google Scholar]
- 14. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jain M (2011) Next-generation sequencing technologies for gene expression profiling in plants. Briefings in Functional Genomics 11: 63–70. 10.1093/bfgp/elr038 [DOI] [PubMed] [Google Scholar]
- 16. Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, et al. (2010) De novo assembly and analysis of RNA-seq data. Nature methods 7: 909–912. 10.1038/nmeth.1517 [DOI] [PubMed] [Google Scholar]
- 18. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics 10: 57–63. 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research 36: 3420–3435. 10.1093/nar/gkn176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome biology 3: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bunsupa S, Yamazaki M, Saito K (2012) Quinolizidine alkaloid biosynthesis: recent advances and future prospects. Frontiers in plant science 3:239 10.3389/fpls.2012.00239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Z, Fang B, Chen J, Zhang X, Luo Z, Huang L, et al. (2010) De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 11: 726 10.1186/1471-2164-11-726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krishnamoorthy B, Parthasarathy V (2010) Improvement of black pepper. CAB Reviews: Perspectives in Agriculture, Veterinary Science, Nutrition and Natural Resources 5: 1–12. [Google Scholar]
- 24. Jose J, Sharma AK (1985) Structure and behaviour of chromosomes in Piper and Peperomia (family Piperaceae). Cytologia 50: 301–310. [Google Scholar]
- 25. Vasavirama K, UPENDER M (2014) PIPERINE: A VALUABLE ALKALOID FROM PIPER SPECIES. International Journal of Pharmacy & Pharmaceutical Sciences 6: 34–38. [Google Scholar]
- 26. Gordo SM, Pinheiro DG, Moreira EC, Rodrigues SM, Poltronieri MC, Lemos OF, et al. (2012) High-throughput sequencing of black pepper root transcriptome. BMC plant biology 12: 168 10.1186/1471-2229-12-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joy N, Asha S, Mallika V, Soniya EV (2013) De novo Transcriptome Sequencing Reveals a Considerable Bias in the Incidence of Simple Sequence Repeats towards the Downstream of ‘Pre-miRNAs’ of Black Pepper. PLoS One 8: e56694 10.1371/journal.pone.0056694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P (2011) Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC bioinformatics 12: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nature Reviews Genetics 12: 671–682. 10.1038/nrg3068 [DOI] [PubMed] [Google Scholar]
- 30. Shi Y, Yan X, Zhao P, Yin H, Zhao X, Xiao H, et al. (2013) Transcriptomic Analysis of a Tertiary Relict Plant, Extreme Xerophyte Reaumuria soongorica to Identify Genes Related to Drought Adaptation. PLoS One 8: e63993 10.1371/journal.pone.0063993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Long Y, Zhang J, Tian X, Wu S, Zhang Q, Zhang J, et al. (2014) De novo assembly of the desert tree Haloxylon ammodendron (CA Mey.) based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics 15: 1111 10.1186/1471-2164-15-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou X, Qian K, Tong Y, Zhu JJ, Qiu X, Zeng X (2014) De Novo Transcriptome of the Hemimetabolous German Cockroach (Blattella germanica). PLoS One 9: e106932 10.1371/journal.pone.0106932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu D, Sui S, Ma J, Li Z, Guo Y, Luo D, et al. (2014) Transcriptomic Analysis of Flower Development in Wintersweet (Chimonanthus praecox). PLoS One 9: e86976 10.1371/journal.pone.0086976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible W-R (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiology 139: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, et al. (1999) Housekeeping genes as internal standards: use and limits. Journal of biotechnology 75: 291–295. [DOI] [PubMed] [Google Scholar]
- 36. Tu L, Zhang X, Liu D, Jin S, Cao J, Zhu L, et al. (2007) Suitable internal control genes for qRT-PCR normalization in cotton fiber development and somatic embryogenesis. Chinese Science Bulletin 52: 3110–3117. [Google Scholar]
- 37. Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnology letters 26: 509–515. [DOI] [PubMed] [Google Scholar]
- 38. Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 20: 176–183. [DOI] [PubMed] [Google Scholar]
- 39. Arcaro CA, Gutierres VO, Assis RP, Moreira TF, Costa PI, Baviera AM, et al. (2014) Piperine, a Natural Bioenhancer, Nullifies the Antidiabetic and Antioxidant Activities of Curcumin in Streptozotocin-Diabetic Rats. PLoS One 9: e113993 10.1371/journal.pone.0113993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bunsupa S, Katayama K, Ikeura E, Oikawa A, Toyooka K, Saito K, et al. (2012) Lysine decarboxylase catalyzes the first step of quinolizidine alkaloid biosynthesis and coevolved with alkaloid production in leguminosae. The Plant Cell 24: 1202–1216. 10.1105/tpc.112.095885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wink M (1987) Quinolizidine alkaloids: biochemistry, metabolism, and function in plants and cell suspension cultures. Planta Med 53: 509–514. [DOI] [PubMed] [Google Scholar]
- 42. Wink M, Hartmann T (1979) Cadaverine-pyruvate transamination: The principal step of enzymatic quinolizidine alkaloid biosynthesis in Lupinus polyphyllus cell suspension cultures. FEBS letters 101: 343–346. [DOI] [PubMed] [Google Scholar]
- 43. Okwute SK, Egharevba HO (2013) Piperine-Type Amides: Review of the Chemical and Biological Characteristics. International Journal of Chemistry 5: 99–122. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLS)
(XLSX)
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.