

Abstract
Signalling through the Hippo (Hpo) pathway involves a kinase cascade, which leads to the phosphorylation and inactivation of the pro-growth transcriptional co-activator Yorkie (Yki). Despite the identification of a large number of pathway members and modulators, our understanding of the molecular events that lead to activation of Hpo and the downstream kinase Warts (Wts) remain incomplete. Recently, targeted degradation of several Hpo pathway components has been demonstrated as a means of regulating pathway activity. In particular, the stability of scaffold protein Salvador (Sav), which is believed to promote Hpo/Wts association, is crucially dependent on its binding partner Hpo. In a cell-based RNAi screen for ubiquitin regulators involved in Sav stability, we identify the HECT domain protein Herc4 (HECT and RLD domain containing E3 ligase) as a Sav E3 ligase. Herc4 expression promotes Sav ubiquitylation and degradation, while Herc4 depletion stabilises Sav. Interestingly, Hpo reduces Sav/Herc4 interaction in a kinase-dependent manner. This suggests the existence of a positive feedback loop, where Hpo stabilises its own positive regulator by antagonising Herc4-mediated degradation of Sav.
Introduction
The Hippo (Hpo) pathway has emerged as a major regulator of growth and tissue size [1,2]. It was originally identified in Drosophila using genetic screens for mutations resulting in tissue overgrowth [3–12]. The core of the Hpo pathway in Drosophila comprises two kinases, Hpo and Warts (Wts). Activated Hpo phosphorylates the downstream kinase Wts, which leads to the Wts-mediated phosphorylation of the transcriptional co-activator Yorkie (Yki) [7,13]. Phosphorylated Yki is sequestered in the cytosol by 14-3-3 proteins, thereby preventing activation of Yki target genes [14,15]. In the nucleus, Yki associates with a number of transcription factors, such as the TEA (Transcription enhancer factor) domain-containing protein Scalloped (Sd) [16,17], to induce the expression of growth-promoting genes (e.g. myc, bantam and diap1) [13,18–20]. Hence, inactivation of Yki by Wts inhibits growth through the transcriptional repression of these pro-growth regulators. The Hpo pathway is highly conserved between mammals and Drosophila [1,2]. In humans, the Hpo orthologues MST1/2 (Mammalian Sterile 20-like) phosphorylate the Wts orthologues LATS1/2 (Large Tumor Suppressor), leading to the phosphorylation of the Yki orthologues YAP and TAZ [21–25].
Over the last decade, various genetic screens and proteomic approaches have identified many new components of the pathway [26]. This has generated a vast amount of information and increased understanding of the molecular mechanisms involved in the regulation of this pathway, leading to the notion that Hpo signalling acts as a sensor for mechanical cues, apico-basal and planar polarity and G-protein coupled receptors [1,2,21,27,28]. Thus, Hpo signalling is a key effector of tissue architecture in growth control.
However, despite the ever-expanding complexity of Hpo upstream signalling, the regulation and function of the core components still remain incompletely understood. In particular, apart from phosphorylation, the role of covalent protein modifications, such as ubiquitylation and SUMOylation has been little explored. One well-characterised instance is the regulation of the Yki orthologs YAP and TAZ by the SCFSlimb/β-TrCP E3 ligase complex [29,30]. Sequential phosphorylation of a YAP/TAZ phospho-degron by Casein Kinase 1∂/ε and LATS1/2 leads to recognition and degradation by SCFSlimb/β-TrCP. Moreover, several other components of the Hpo pathway have been reported to be targets of ubiquitin-dependent proteasomal degradation, such as RASSF1A, RASSF5/NORE1, Mob1 and Wts/LATS1 [31–36].
The present study focuses on the tumor suppressor protein Salvador (Sav), which, together with Hpo and Wts, was one of the first Hpo pathway components to be identified [5,6]. Sav is a WW domain-containing scaffold protein, which interacts with the Hpo kinase via their C-terminal SARAH (Salvador, RASSF, Hippo) domains [5,6,37]. The functional importance of Sav/Hpo dimerisation for Hpo kinase activity remains unclear, since Hpo/MST dimerisation is sufficient to promote activation by trans-phosphorylation [38–40]. However, genetic evidence in flies and vertebrates clearly indicates that Sav is required for Hpo pathway activity. For instance, loss of fly sav phenocopies the overgrowth phenotype of hpo mutants [5,6,41,42], while knockout mice for WW45/Sav1 display YAP-dependent phenotypes in multiple tissues [9,41–43]. Recent work indicates that Sav promotes membrane recruitment of Hpo, where it can phosphorylate and activate Wts [44]. Another clear consequence of Sav/Hpo dimerization is the phosphorylation and stabilization of Sav, both in flies and mammals [7,9,45,46]. However, it is not clear whether Sav phosphorylation by Hpo is responsible for its stabilization, since a point mutant in the ATP-binding pocket (Lysine to Arginine, K/R) of Hpo/MST still stabilizes Sav [9,45]. Here, we use an RNAi screening approach to identify regulators of Sav stability. We identify the HECT E3 ubiquitin ligase Herc4 as a negative modulator of Hpo signalling. In addition, we show that Hpo can stabilise Sav by interfering with Herc4-dependent Sav degradation.
Material and Methods
Drosophila cell culture and expression constructs
Drosophila S2 cells were transfected with Effectene transfection reagent (Qiagen) and grown in Drosophila Schneider’s medium (Life Technologies) containing 10% FBS (Sigma), 50μg/ml penicillin and 50 μg/ml streptomycin. Expression plasmids were generated using Gateway® technology (Life Technologies). Open reading frames (ORF) were PCR amplified from existing plasmids or cDNA (DGRC, https://dgrc.cgb.indiana.edu/vectors/) and cloned into Entry vectors (pENTRTM/D-TOPO). Vectors from the Drosophila Gateway Vector Collection (http://www.ciwemb.edu/labs/murphy/Gateway%20vectors.html) were used as Destination vectors. All expression plasmids were sequence-verified. Point mutations were generated using the Quikchange® Multi Site-Directed Mutagenesis Kit (Stratagene).
dsRNA production and treatment
RNAi library generation work-plan can be found in the supplementary figures (S2–S4 Figs). dsRNAs were synthesized using the Megascript T7 kit (Ambion). DNA templates for dsRNA synthesis were PCR-amplified from genomic DNA using primers that contained the 5’ T7 RNA polymerase-binding site sequence. dsRNA primers were designed using the DKFZ RNAi design tool (http://www.dkfz.de/signaling2/e-rnai/)
RNAi screening was carried out as outlined in S3 Fig. Follow-up RNAi experiments were carried out in 6 well plates using S2 cells. 1-2x106 cells were plated per well and cells left to settle. After 3 h the medium was removed and replaced with 1 ml of serum free Schneider’s medium containing dsRNAs (20 μg). Cells were soaked for 30 min and then 2 ml of full Schneider’s medium was added. Transfections were carried out 24 h after dsRNA treatment.
Immunoprecipitation and immunoblot analysis
For immunoprecipitation assays, cells were lysed in Lysis Buffer (50 mM Tris pH8, 150 mM NaCl, 1% NP-40, and 1 mM EGTA) supplemented with 0.1M NaF, phosphatase inhibitor cocktails 2 and 3 (Sigma) and protease inhibitor cocktail (Roche). Cell extracts were cleared at 16,000 g for 20 min at 4°C. FLAG-tagged proteins were purified using anti-FLAG M2 Affinity Agarose Gel (Sigma), whereas immunoprecipitation of Myc-tagged proteins was carried out using Protein A Sepharose (Sigma). Detection of purified proteins and associated complexes was performed by immunoblot analysis using chemiluminescence (Pierce). Western Blots were probed with mouse anti-FLAG (M2, Sigma), rabbit anti-FLAG (Sigma), mouse anti-Myc (9E10, Santa Cruz Biotechnology), rabbit anti-Myc (Santa Cruz Biotechnology), rat anti-HA (3F10, Roche Applied Science), mouse anti-Tubulin (E7, Developmental Studies Hybridoma Bank, DSHB) and mouse anti-GFP (in-house produced), rat anti-Yki [47], and rabbit [48] and guinea pig anti Sav (a kind gift from G. Halder).
Ubiquitylation assays
For ubiquitylation assays S2 cells were transfected with Myc-Sav and HA-Ubiquitin and, unless otherwise stated, treated with proteasome inhibitors for 4 h. Cells were harvested and lysed in Lysis buffer (see above). Lysates were immunoprecipitated with an anti-Myc antibody and Protein A Sepharose (Sigma), and subjected to western blot analysis. Ubiquitylation was detected using an antibody against HA-Ubiquitin. Lysis buffer containing 5mM N-Ethylmaleimide (NEM) (Sigma) was used to block deubiquitylating enzyme activity.
Proteasome inhibitor treatment
For proteasome inhibition experiments, cells were treated with 50μM MG132 (Calbiochem) and 50μM calpain inhibitor I (Ac-LLnL-CHO or LLnL) (Sigma) for 4 h before cell lysis. Ubiquitylation assays were carried out after proteasome inhibitor treatment, unless stated otherwise.
Western blot analysis of fly heads
Heads were cut off from frozen animals and lysed in RIPA buffer (10 mM Tris HCl pH 7.5, 150 mM NaCl, 1% (v/v) Triton X-100, 0.1% (w/v) SDS, 1% (w/v) Sodium deoxycholate) supplemented with 0.1M NaF, phosphatase inhibitor cocktails 2 and 3 (Sigma) and protease inhibitor cocktail (Roche). Tissue lysates were incubated on ice for 10 min and subsequently cleared by centrifugation at 16,000 g for 10 min at 4°C. 150ug of total lysate was loaded per lane.
Immunofluorescence
Wing imaginal discs from third-instar larvae were dissected, fixed for 20 min in 4% formaldehyde, washed three times in PBS supplemented with 0.1% Triton X-100 (PBS-T) and pre-blocked in PBS supplemented with 0.3% Triton X-100 and 10% normal goat serum (NGS) for 1 h. Discs were incubated overnight in the primary antibody diluted in PBS-T containing 10% NGS, followed by three washes with PBS-T, and incubation with a secondary antibody in PBS-T/10% NGS for 2 h at room temperature. After three further washes, discs were mounted in Vectashield mounting medium with DAPI (Vectorlabs). Rat anti-Ci155 antibody (Developmental Studies Hybridoma Bank, monoclonal 2A1) was used at 1:20. Secondary anti-rat Alexa 647 antibody (Molecular Probes) was used at 1:200. Fluorescence images were acquired on a Zeiss LSM710 confocal laser scanning microscope (40x objective lens).
Sequence Alignments
Multiple sequence alignment and phylogenetic tree analysis was carried out using Clustal Omega (EMBL-EBI). Protein sequence accession numbers were as follows: Itch hs BAB39389.1 (GenBank), Nedd4 hs AAI44285 (AAI44285), Smurf1 hs EAL23885 (EAL23885), Herc1 hs Q15751.2 (UniProtKB/Swiss-Prot), Herc2 hs O95714.2 (UniProtKB/Swiss-Prot), Herc3 hs Q15034.1 (UniProtKB/Swiss-Prot), Herc4 hs Q5GLZ8.1 (UniProtKB/Swiss-Prot), Herc5 hs Q9UII4.2 (UniProtKB/Swiss-Prot), Herc6 hs Q8IVU3.2 (UniProtKB/Swiss-Prot), Herc4 dm (CG9153) Q5GLZ8.1 (UniProtKB/Swiss-Prot).
Generation of herc4 CRISPR mutant
Introduction of a frame-shift mutation in the herc4 gene was performed using the CRISPR/Cas9 system [49]. Suitable target sites for guide RNAs (sgRNA) were determined using http://www.flyrnai.org/crispr/. Sense and anti-sense oligos of the selected sgRNA were annealed, phosphorylated and cloned into the expression vector pCFD3 according to the protocol described on http://www.crisprflydesign.org/grna-expression-vectors/. DNA was injected into embryos of y 1, M{nos-Cas9.P}ZH-2A w* transgenic flies (Bloomington stock number 54591) by the Fly Facility at the University of Cambridge Department of Genetics. G0 flies were then crossed with the w;;TM3/TM6 balancer strain; subsequently, G1 male flies were crossed with the same balancer strain, followed by genomic DNA screening for frame-shift mutations. We also balanced a wild type progeny of the injected founders to use as a control strain (herc4 ctrl).
Analysis of Drosophila adult wings
Adult wings were mounted using Euparal mounting medium (Agar Scientific) and imaged using a Zeiss Axio-plan 2 imaging system, using Optovar optics (1.6x) and a 2.5x objective (Plan-NEOFLUAR, Zeiss) connected to a Leica DFC420C camera. Wings were subsequently measured and quantified using ImageJ.
Genotypes
Fig 6: left panels: w; ubi-GFP::sav/+; hh-GAL4/+, right panels: w; ubi-GFP::sav/+; hh-GAL4/UAS-Herc4.
Fig 6. Herc4 overexpression reduces Sav protein levels in vivo.
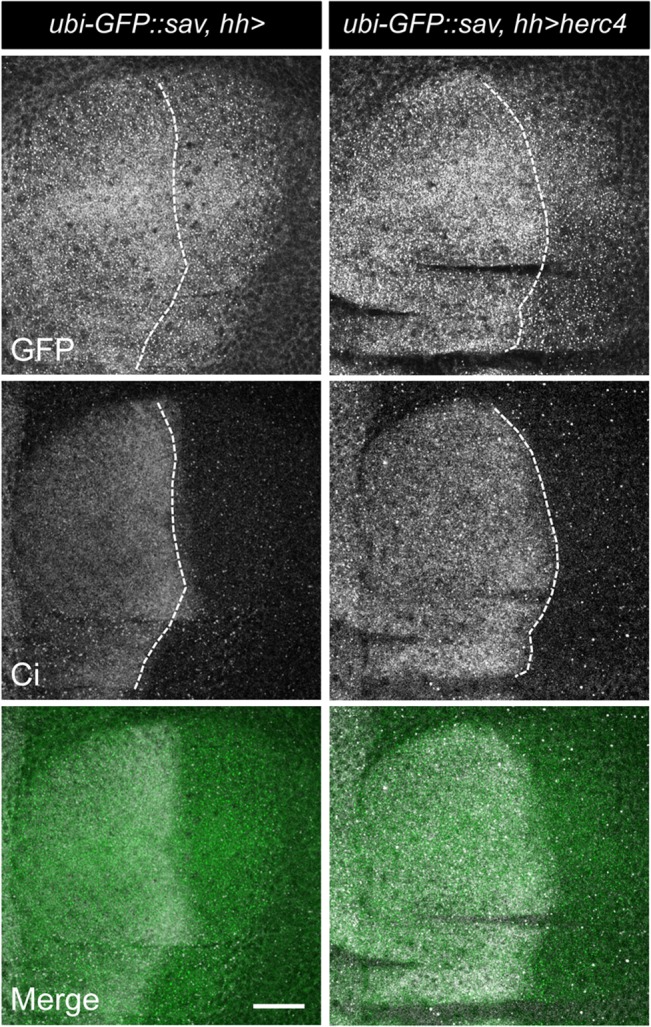
GFP-tagged Sav was expressed under the control of the ubiquitin promoter (ubi-GFP::Sav) and analysed in 3rd instar larval wing discs expressing hh-GAL4 alone (left panel) or UAS-herc4 in the posterior compartment (hh>herc4). White dotted lines denote the border between the anterior and posterior compartments of the wing. Anti-Ci staining marks the anterior compartment. Scale bar = 100μm.
Fig 10: w; UAS-CD8-GFP; rn-GAL4. w; UAS-herc4; rn-GAL4., w; UAS-herc4-C/A; rn-GAL4. w; UAS-CD8-GFP; rn-GAL4, UAS-sav. w; UAS-herc4; rn-GAL4, UAS-sav. w; UAS-herc4-C/A; rn-GAL4, UAS-sav. w; UAS-CD8-GFP; rn-GAL4, UAS-sav-S413A. w; UAS-sav-S413A; rn-GAL4, UAS-herc4. w; UAS-herc4-C/A; rn-GAL4, UAS-sav-S413A.
Fig 10. Growth regulatory function of Herc4.

(A) Overexpression of Herc4 leads to a partial rescue of the Sav overexpression phenotype. Expression of sav (rn>sav) and savS413A (rn>savS413A) transgenes under the control of the rotund (rn) promoter causes tissue undergrowth and results in smaller wings. Both transgenes were combined with either UAS-herc4, UAS-herc4C/A or UAS-CD8-GFP to address the effect of Herc4. (B) Quantification of wing sizes. Shown are fold changes of average wing areas for the indicated genotypes compared to the control (rn>UAS-CD8-GFP), which was set as 1. Statistical significance assessed by Student’s t-test. * p < 0.0001. N.S. p > 0.05. n = 10–21. Error bars denote standard deviations.
Fig 11: w;; rn-GAL4/ctrl. w;; rn-GAL4, UAS-sav/ctrl. w;; rn-GAL4, UAS-sav-S413A/ctrl. w;; rn-GAL4/herc4 C6.3. w;; rn-GAL4, UAS-sav/ herc4 C6.3. w;; rn-GAL4, UAS-sav-S413A/ herc4 C6.3.
Fig 11. Generation and in vivo analysis of herc4 C6.3 mutant.

(A) Sequence analysis of herc4 CRISPR mutation. Sequence chromatogram of the sgRNA target sequence (boxed in the sequence alignment on the right) showing the loss of cytosine 92 (C92) in the herc4 C6.3 strain (marked by asterisk). (B) Diagram and sequence of the predicted protein resulting from frame-shift mutation. The loss of C92 results in a frame-shift mutation and a truncated protein of 30aa plus 24aa of out-of-frame sequence. (C) herc4 C6.3 enhances the Sav overexpression phenotype in the wing. Both sav (rn>sav) and savS413A (rn>savS413A) transgenes were combined with heterozygosity for either herc4 C6.3 or herc4 ctrl and wings sizes were analysed. (D) Quantification of wing sizes. Shown are fold changes of average wing areas for the indicated genotypes compared to the control (rn/ctrl), which was set as 1. Statistical significance assessed by Student’s t-test. * p < 0.0001. N.S. p > 0.05. n = 10–21. Error bars denote standard deviations. (E) Levels of Sav protein in fly heads of herc4 C6.3, herc4 ctrl and GMR>savS413A were analysed by Western blot. Bottom panel: quantification presented as ratio of Sav/Yki levels from three independent repeats. * p = 0.0385 with a Student’s t-test.
Results
Sav is ubiquitylated and degraded by the proteasome
Sav is an unstable protein and its stability is regulated by the proteasome system (Fig 1A , [7,9,45,46]). As proteasomal degradation is triggered by poly-ubiquitylation of target proteins, we performed a cell-based ubiquitylation assay by co-expressing Myc-tagged Sav with HA-tagged ubiquitin, followed by Myc immunoprecipitation (Fig 1A ). This revealed the presence of Sav poly-ubiquitylated species, which were stabilised by treatment with proteasome inhibitors. Ubiquitin conjugation primarily occurs through the formation of an isopeptide bond between the ubiquitin C-terminus and the epsilon amino group of lysine residues on the substrate [50]. Sav contains eighteen lysine residues, clustered at both termini of the protein, ten are contained within amino acids 12 to 57 and eight are located in the area containing the conserved WW and SARAH domains (Fig 1B ). To establish the domain where Sav ubiquitylation occurs, we analysed Sav truncations spanning either the N or C terminal part of the protein (Sav∆C and Sav∆N). As shown in Fig 1B , Sav ubiquitylation occurs within the conserved C-terminal domain, comprising the WW and SARAH domain.
Fig 1. Sav is ubiquitylated predominantly in the C-term half of the protein.
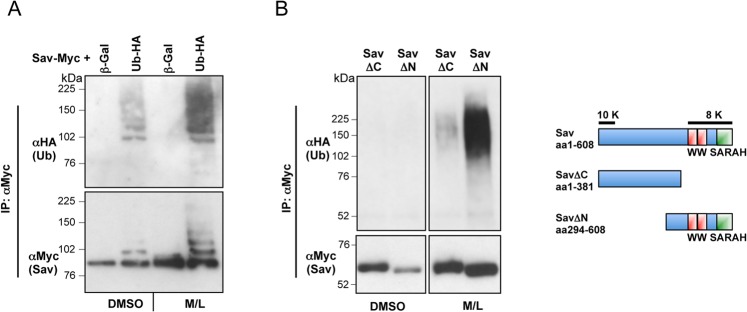
(A) Sav is stabilised and shows increased ubiquitylation in the presence of proteasome inhibitors (MG132 and LLnL). Myc-tagged Sav was expressed in S2 cells with either Ub-HA or ß-Galactosidase (ß-Gal) in the presence of proteasome inhibitors (MG132 and LLnL, M/L) or DMSO control. Sav ubiquitylation assay shows incorporation of HA-tagged ubiquitin. (B) Left panel: Sav ubiquitylation occurs in the C-terminal half of the protein. Sav deletions (∆C and ∆N) were tested for ubiquitylation as described above. Right panel: schematic representation of Sav protein structure and distribution of Lysine residues. Sav contains eighteen Lysine residues (K), which could serve as potential ubiquitylation sites: ten are located near the N-terminal end of the protein, whereas the remaining eight are located in the C-terminus.
Hpo stabilises Sav by inhibiting its ubiquitylation
We and others have observed that exogenous Sav is stabilised by Hpo co-expression, while Hpo depletion causes endogenous Sav downregulation (Fig 2A and 2B , [7,9,45,46,48]). In order to further investigate the role of Hpo in regulating Sav stability, we carried out ubiquitylation assays using full-length Sav or Sav truncations (Sav∆C and Sav∆N) in the presence or absence of Hpo. Co-expression of Hpo resulted in a reduction in Sav ubiquitylated species (Fig 2C ), indicating that Hpo regulates Sav stability by preventing its ubiquitylation.
Fig 2. Hpo stabilises Sav by inhibiting Sav ubiquitylation.

(A) Sav is stabilised when co-expressed with Hpo. Baseline Sav expression is increased upon proteasome inhibitor treatment (M/L). (B) hpo depletion by RNAi destabilises Sav protein levels. Top panel: endogenous Sav levels (arrow) in non-transfected S2 cells. Asterisk indicates a non-specific band detected by the anti-Sav antibody. Bottom panel: levels of transfected Myc-tagged Sav. Sav depletion by RNAi is used as a control. (C) Hpo inhibits ubiquitylation of Sav. Sav, Sav∆N, and Sav∆C were analysed for ubiquitylation in the presence or absence of co-expressed Hpo. Hpo prevents ubiquitylation of full-length Sav and Sav∆N, while Sav∆C is not ubiquitylated. (A-C) S2 cells were transfected with the indicated plasmids or treated with the indicated dsRNAs prior to lysis. Lysates were processed for western blot analysis or used in co-IP.
Since Sav ubiquitylation occurs in the C-terminal domain responsible for Hpo binding, Hpo could sterically interfere with ubiquitin ligase binding or ubiquitin transfer. Alternatively, since Hpo is known to phosphorylate Sav ([7,9,45,46,51] and this report), phosphorylation might prevent ligase binding or ubiquitylation. To investigate these possible mechanisms, we analysed the effect of several Hpo truncations and mutants on Sav ubiquitylation (Fig 3A ). Hpo constructs that bind but fail to phosphorylate Sav, either by deletion of the kinase domain (Hpo∆N) or point mutation in the T-loop domain (at residue 195 –HpoT195A), were unable to prevent Sav ubiquitylation (Fig 3B ). Similarly, we tested two C-terminal truncations of Hpo lacking the SARAH domain but still containing the Hpo PPxY motif, the longer of which still binds Hpo robustly (Hpo∆C2), while the shorter truncation is reduced in its ability to bind Hpo (Hpo∆C3), as well as a version of Hpo∆C3 carrying a mutation in the PPxY motif (Hpo∆C3/Y591G) motif, which cannot bind Sav at all (Fig 3C ). These experiments showed that the strength of Hpo/Sav binding correlates with the effect of Hpo on Sav ubiquitylation, with Hpo∆C2 strongly blocking Sav ubiquitylation, and Hpo∆C3 and Hpo∆C3/Y591G having increasingly weaker effects (Fig 3C ). These results indicate that Hpo requires both Sav binding and kinase activity to inhibit Sav ubiquitylation, thereby excluding steric interference as the sole mechanism for Hpo action.
Fig 3. Binding of active Hpo to Sav is required to inhibit Sav ubiquitylation.

(A) Schematic representation of Hpo constructs used. Protein domains and point mutations in the Hpo T-loop inactivating Hpo kinase (HpoT195A) and PPxY motif (Y591G) are indicated. (B) Sav ubiquitylation assays with co-expressed Hpo truncations (Hpo∆N and ∆C) or Hpo kinase dead (HpoT195A). While full-length Hpo prevents Sav ubiquitylation, truncations lacking the kinase domain or C-terminus, or a kinase-dead Hpo have no effect. (C) Hpo C-terminal deletions (Hpo∆C2 and Hpo∆C3) and a Hpo C-terminal deletion mutated for the PPxY motif (Hpo∆C3/Y591G) affect Sav ubiquitylation proportionally to their ability to bind Sav (compare upper and lower panels).
A ubiquitin ligase RNAi screen identifies Herc4 as a Sav E3 ligase
To identify proteins responsible for Sav ubiquitylation, we carried out a cell-based RNAi screen using a Drosophila dsRNA sub-library containing known and candidate genes involved in ubiquitylation (378 genes involved in ubiquitin conjugation and ubiquitin binding). The effect of dsRNAs on Sav-Myc expression was analyzed by standard Western-blot analysis. Using this approach we identified the HECT domain protein Herc4 (CG9153) as a Sav E3 ligase (Fig 4A ). Drosophila Herc4 contains a RLD (RCC1-like domains) domain comprising 7 RCC1-like repeats and a HECT domain [52] (Fig 4B ). RLD domains are structural scaffolds with a β-propeller architecture believed to be involved in protein-protein interactions, whereas the HECT domain is a conserved E3 ligase domain [53,54]. HECT domains bind specific ubiquitin-conjugating enzymes (E2s), accept ubiquitin from the E2 to form a ubiquitin-thioester intermediate with the HECT active cysteine (Fig 4B ), then transfer ubiquitin to the substrate. Herc4 belongs to the subfamily of small Herc E3 ligases, which in humans comprises 4 members, Herc3-6 [52]. In Drosophila there are only 2 small Herc E3 ligases, Herc2 and Herc4. Drosophila Herc4 is marginally more related to human Herc4 than human Herc3 (45% and 44% identity between CG9153 and hsHerc4 and hsHerc3, respectively; Fig 4C ).
Fig 4. Ubiquitin ligase RNAi screen identifies Herc4 as a Sav E3 ligase.

(A) E3 RNAi screen work-plan (B) Diagram depicting the Herc4 protein domain structure and alignment of HECT domains. Herc4 contains seven RCC1 repeats and a HECT E3 ligase domain. Protein alignment shows the last 36 aa of Drosophila melanogaster Herc4 aligned with the corresponding sequences of human Nedd4, Itch, Smurf, and Herc3. The conserved catalytic cysteine is shown in red. (C) Phylogenetic tree of human Herc1-6 and Drosophila melanogaster Herc4.
Herc4 controls Sav stability and ubiquitylation in vitro and in vivo
HECT E3 ligases physically interact with their substrates [54]. To determine whether Herc4 associates with Sav, we performed co-immunoprecipitation (co-IP) experiments in S2 cells using tagged Herc4 and Sav. Herc4 robustly associated with Sav, but not with a control protein, Rep3 (Fig 5A ). We identified Herc4 based on the ability of herc4 RNAi to increase Sav protein levels in S2 cells (Fig 5B ). To further investigate the functional significance of this finding, the role of Herc4 in Sav ubiquitylation was analysed. We performed a cell-based ubiquitylation assay in the presence of the deubiquitylating enzyme inhibitor NEM to preserve ubiquitylated species. As shown in Fig 5C , herc4 RNAi resulted in a marked reduction of Sav ubiquitylation compared to control RNAi treatment, indicating that Herc4 is required for Sav ubiquitylation.
Fig 5. Herc4 controls Sav stability and ubiquitylation.

(A) Sav binds to Herc4. Myc-tagged Sav was immunoprecipitated using a Myc antibody and bound Herc4-Flag protein detected using Flag antibody. Myc-tagged Drosophila Rep3 was used as control. (B) herc4 RNAi stabilises Sav protein levels. Left panel: endogenous Sav levels (arrow) in non-transfected S2 cells. Asterisk indicates a non-specific band detected by the anti-Sav antibody. Left bottom panel: quantification presented as ratio of Sav/Tub levels. Right panel: levels of transfected Myc-tagged Sav. Right bottom panel: quantification presented as ratio of Sav-Myc/Tub levels (C) herc4 RNAi reduces Sav ubiquitylation. Sav ubiquitylation assay of Myc-tagged Sav expressed in S2 cells, treated with dsRed RNAi, herc4 RNAi or sav RNAi. Ubiquitylation assay was performed in the presence of 5mM NEM to block deubiquitylating enzyme activity (D) Overexpression of Herc4 results in destabilisation and increased ubiquitylation of Sav, which is dependent on the active site cysteine. Sav ubiquitylation assay of Myc-tagged Sav, Sav∆N and Sav∆C co-expressing either Flag-Herc4 or Flag-Herc4C/A.
To assess the specificity of Herc4 as a Sav E3 ligase, we analyzed the effect of Herc4 overexpression on Sav ubiquitylation. Expression of Herc4 enhanced Sav ubiquitylation and reduced Sav protein levels in cell lysates, while a catalytically impaired Cysteine to Alanine mutant (Herc4C/A) had a reduced effect (Fig 5D ). In agreement with our previous observations (Fig 1B ), the C-terminal portion of Sav (Sav∆N), but not the N-terminal (Sav∆C), binds to and is ubiquitylated by Herc4 (Fig 5D ). In contrast, two other HECT domain E3 ligases, Su(dx) and Nedd4, did not promote Sav ubiquitylation (S1 Fig).
Next, we explored the effect of Herc4 on Sav stability in vivo. We generated a GFP-tagged sav transgene, expressed under the control of the ubiquitin promoter (ubi-GFP::sav). Using the GAL4/UAS system, UAS-Herc4 was expressed in the posterior compartment of wing imaginal discs, the larval precursor to the adult wing, using the hh-Gal4 (referred to as hh>herc4) driver. Cubitus interruptus (Ci) staining was used to differentiate the anterior (Ci-positive) and posterior (Ci-negative) compartments of the wing imaginal disc. Herc4 overexpression caused depletion of Sav levels (Fig 6, right panels), indicated by the reduction in the GFP signal. These results are consistent with our biochemical data showing that Herc4 promotes Sav ubiquitylation and proteasomal degradation.
Mapping the Herc4/Sav interaction domains
To map the domains required for the interaction between Herc4 and Sav, we carried out co-IP experiments using fragments of either protein (Fig 7). Using this strategy, we were able to map the Herc4 interaction domain to the RLD domain as deleting the whole RLD abolishes binding between the two proteins. Deletion of RCC1-like repeats 1–3 had no effect on binding, neither did deletion of the entire HECT domain (Fig 7A ). Thus, Herc4 binds Sav via RCC1-like domains 4–7.
Fig 7. Mapping of Herc4/Sav binding.

(A) Mapping of Herc4 interaction domain with Sav. Co-IPs of Sav with Herc4 truncations lacking the N-terminal RCC1 domains (∆RCC1-3, ∆RCC1-7) or the HECT domain (∆HECT). The RCC4-7 repeats are required for binding of Herc4 to Sav. (B) Mapping of the Sav interaction domain with Herc4. C-terminal fragments of Sav were used in co-immunoprecipitation experiments with Herc4. (C) Schematic representation of Herc4 and Sav truncations used in A and B.
Herc4 binds to the conserved C-terminus of the Sav protein (Fig 5D ). To precisely define the region responsible for Sav/Herc4 binding, we analysed C-terminal fragments of Sav in co-IP experiments with Herc4 (Fig 7B ). The domain responsible for the association of Herc4 to Sav can be narrowed down to the region stretching from the end of the WW domains to the beginning of the SARAH domain (aa 492–546), as deleting this domain abolishes binding between Herc4 and Sav (Fig 7B ). Adding the SARAH domain (Sav∆N4) renders the interaction more robust, and elicits the appearance of a ladder of higher molecular species, which could indicate poly-ubiquitylation (Fig 7B ).
Antagonism between Herc4 and Hpo determines Sav stability
As both Herc4 and Hpo have a role in regulating Sav ubiquitylation and stability, we tested a possible functional interaction between the two proteins. Combined expression of Hpo and Herc4 resulted in reduced Herc4-induced Sav ubiquitylation (Fig 8A ). We also noted that upon Hpo co-expression there was reduced association between Herc4 and Sav (Fig 8A ). Similarly, Hpo reduced Sav levels in Herc4 immunoprecipitates (Fig 8B ). Interestingly, the kinase dead version of Hpo (HpoT195A) did not affect Sav/Herc4 binding (Fig 8B ), indicating that the effect of Hpo is phosphorylation-dependent. These results suggest that phosphorylation of Sav by Hpo prevents Herc4/Sav binding, leading to Sav stabilization. Depletion of hpo causes Sav to be destabilized (Fig 2B ), suggesting that under those conditions Sav is no longer protected from Herc4-mediated degradation. Indeed, co-depletion of herc4 and hpo restored Sav levels (Fig 8C ). Thus, the interplay between Hpo and Herc4 determines Sav stability.
Fig 8. Hpo competes with Herc4 for Sav binding and antagonises Herc4-mediated ubiquitylation of Sav.

(A) Hpo reduces Herc4-induced ubiquitylation of Sav. Sav ubiquitylation assay was performed using Myc-tagged Sav co-expressed with either Hpo, Herc4 or Hpo and Herc4 combined. Ubiquitylation assay was performed in the presence of 5mM NEM to block deubiquitylating enzyme activity. (B) Binding of Sav to Herc4 is prevented by active Hpo kinase (compare left with right lane), while kinase-dead Hpo has no effect (compare left and middle lanes). Herc4/Sav co-IP experiments were performed in the presence of a control protein (GFP), wild-type or kinase dead version of Hpo (HpoT195A). (C) herc4 RNAi rescues the destabilizing effect of hpo RNAi on Sav protein levels. S2 cells expressing Myc-tagged Sav (and GFP-Flag as a control) were treated with the indicated combinations of dsRNA.
herc4 depletion prevents Sav destabilization caused by loss of apical Hpo pathway regulators
Recent reports have highlighted the importance of the cell cortex as an organising centre for Hpo pathway activity [44,47,55–58]. Several apically localized proteins, such as the Kibra/Expanded/Merlin (KEM) complex and the immunoglobulin super-family trans-membrane protein Echinoid (Ed), have been proposed to recruit Hpo/Sav to the cell membrane. In particular, Ed has been shown to bind Sav and prevent its degradation [55].
Expression of Ed and Kibra in S2 cells stabilises co-transfected Sav (Fig 9A and [55]). Conversely, loss of either Kibra or Ed causes a marked reduction of Sav levels (Fig 9B and 9C ), suggesting that loss of apical tethering of Sav makes it a target for ubiquitylation and degradation. To investigate this assumption further, we tested the effect of depleting herc4 in this context. S2 cells were transfected with Sav-Myc and treated with dsRNA for kibra and ed in combination with herc4 RNAi or lacZ RNAi as a control. herc4 RNAi rescued the destabilising effect of ed and kibra RNAi (Fig 9B and 9C ). Thus, when apical members of the Hpo pathway are depleted, Sav is released into the cytosol, where Herc4 promotes its ubiquitylation and subsequent degradation by the proteasome.
Fig 9. herc4 depletion prevents the decrease in Sav protein levels elicited by loss of apical Hpo pathway regulators.

(A) Co-expression of Ed and Kibra stabilizes Myc-tagged Sav. Hpo co-transfection is used as a positive control. (B) herc4 RNAi rescues ed RNAi-induced reduction in Sav protein levels. (C) herc4 RNAi rescues kibra RNAi-induced reduction in Sav protein levels. (A-C) S2 cells were transfected with Sav-Myc and the indicated plasmids, and treated with the indicated dsRNAs before lysis and western blot analysis.
Growth regulatory function of Herc4
One of the main effects of Hpo pathway disruption in vivo is an increase in tissue size. To investigate the impact of Herc4 on tissue growth control via Sav, we combined GAL4/UAS-driven Herc4 expression with transgenic lines expressing either wild type Sav or a version which is refractory to inhibition by the Salt-Inducible Kinases (Sik2/3) and therefore more active (SavS413A) [59]. The transgenes were expressed under the control of the wing-specific driver rotund-Gal4 (rn-Gal4). Overexpression of wild type Sav modestly reduced the size of the wing, while the SavS413A mutant had a stronger effect (Fig 10A ). Expression of UAS-herc4 was sufficient to partially suppress the undergrowth phenotype caused by Sav overexpression. This effect was dependent on the E3 ligase activity of Herc4, since overexpression of a HECT mutant version of Herc4 (UAS-Herc4C/A) had no effect on wing size (Fig 10A and 10B ). Thus, Herc4 expression is able to suppress Sav growth-inhibitory function in vivo.
To study the effect of loss of Herc4 activity we generated a herc4 mutant using CRISPR technology [49]. Using an sgRNA that targets the second exon of herc4, we generated a mutation, herc4 C6.3, carrying a one base-pair deletion at codon position C92 (cysteine 92) (Fig 11A ). This mutation induces a frame-shift resulting in the production of a 30aa peptide (followed by 24aa of out-of-frame sequence), which lacks most of the Herc4 protein (Fig 11B ). Homozygous herc4 C6.3 mutant animals were viable and fertile and had no apparent morphological abnormalities. This is not surprising, as modest overexpression of Sav, for instance in the eye under the GMR promoter, does not lead to a visible phenotype [5]. We therefore tested whether herc4 C6.3 could modify the undergrowth phenotype elicited by strong overexpression of Sav in the wing under the rn-GAL4 driver (Fig 11C and 11D ). Interestingly, loss of herc4 C6.3 does not modify the undergrowth induced by savS413A, which is already an activated form. By contrast, the herc4 C6.3 mutant significantly enhanced the undergrowth induced by wild type Sav overexpression.
To test whether loss of herc4 affects Sav protein levels in vivo, we analysed Sav protein levels in fly heads by Western blot. Fig 11E shows that Sav protein levels were increased in herc4 C6.3 heads compared to control heads. Together, these results support a role for Herc4 as a Sav E3 ligase.
Discussion
The scaffold protein Sav is a core component of the Hpo signalling pathway and its genetic inactivation is sufficient to increase tissue growth in flies and mice [5,6,41,43]. Sav dimerises with Hpo through their respective SARAH domains [7–11], and recruits Hpo to the plasma membrane, thereby promoting Wts phosphorylation [44]. In addition, Sav associates with the downstream kinase Wts via a WW domain/PPxY interaction [5]. Sav may therefore scaffold the Hpo/Wts core kinase cassette, thereby further strengthening Wts phosphorylation and activation. Indeed, mammalian Sav1/WW45 immunoprecipitates with endogenous Lats1 and Mst2 [21], and Sav strengthens the Hpo/Wts association in Drosophila tissue culture cells [59].
Sav levels appear to be tightly regulated by proteasomal degradation, and its association with Hpo/MST promotes both its phosphorylation and stabilisation ([7,9,45,46], this report). Using a point mutation in the ATP-binding pocket (Lysine to Arginine, K/R), we and others had observed that Hpo/MST kinase activity appeared dispensable for Sav stabilisation, opening the possibility for the involvement of another kinase [9,45]. However, since a version of Hpo carrying a mutation in the T-loop (T195A) and a truncation lacking the kinase domain both fail to phosphorylate and stabilise Sav (Fig 3), it seems most likely that Hpo kinase activity is in fact responsible for stabilising Sav, suggesting that the HpoK/R mutant has residual activity.
In order to shed light on the regulation of Sav stability, we performed a screen for Sav E3 ligases, leading us to identify Herc4 as a negative regulator of Sav (Fig 4). We show that herc4 depletion reduces Sav ubiquitylation in cell culture (Fig 5) and that herc4 mutants have elevated Sav levels in vivo (Fig 11). However, since the increase in Sav levels in herc4 mutants are relatively modest, it is possible that other Sav ubiquitin ligases operate in parallel to Herc4. The Herc family is a poorly characterised group of six proteins containing a C-terminal HECT ubiquitin ligase domain and at least one N-terminal RLD [52]. Mammalian Herc1 and 2 are large (~500 KDa) proteins containing multiple RLDs as well as other domains (WD40, zinc fingers), while Herc3-6 have shorter N-termini containing one RLD domain. As a β-propeller structure, the RLD domain is likely to constitute a substrate-recruitment motif [53]. Indeed, we showed that Sav binding requires RCC1 repeats 4–7 (Fig 7). Drosophila has a single Herc1/2 ortholog (called Herc2) and a single Herc3-6 ortholog (CG9153, which we name Herc4 based on sequence homology). Herc4 mutant mice are viable but males display a spermatozoon maturation defect [60]. In addition, Herc4 was found to be upregulated in over half of a cohort of invasive ductal breast carcinoma [61]. It would therefore be interesting to examine Sav1/WW45 levels in these tumours, as well as assess their differentiation status, since recent work shows that Sav is required for terminal differentiation in the mouse breast epithelium [42].
In this report, we have also shown that the ability of Herc4 to degrade Sav is antagonised by Hpo (Fig 8). Furthermore, Hpo co-expression is able to displace Herc4 from Sav in a kinase domain-dependent manner. This could be due to phosphorylation by Hpo of the interaction surface on either Sav or Herc4. Our data are consistent with a model in which apical recruitment of Sav by proteins such as Ed and the KEM complex can stabilise Sav, perhaps by bringing it in close proximity to active Hpo at the plasma membrane. This would lead to stabilisation of Sav, promote Wts activation by Hpo and, ultimately, lead to Yki inhibition. We therefore propose that Hpo can strengthen the activation of the Hpo pathway core kinase cassette by phosphorylating its own adaptor Sav, thereby preserving it from Herc4-dependent degradation.
Supporting Information
Flag-tagged Herc4, Su(dx) and Nedd4 were expressed in S2 cells together with Sav-Myc and Ub-HA. After lysis, Sav was analysed for ubiquitylation.
(TIF)
(TIF)
(TIF)
(TIF)
Acknowledgments
We are grateful to P. Ribeiro, Y. Zhou and T. Bertran for comments on the manuscript, P. Gaspar for genetics advice, M. Holder for CRISPR advice, P. Meier for advice on the RNAi library, T. Gilbank, S. Maloney, C. Gillen and P. Moulder for fly maintenance and G. Halder for the anti-Sav antibody. We would also like to thank BestGene Inc. and the Fly facility at the University of Cambridge for injection services.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by Cancer Research UK (number 17064).
References
- 1. Yu FX, Guan KL (2013) The Hippo pathway: regulators and regulations. Genes Dev 27: 355–371. 10.1101/gad.210773.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin JI, Poon CL, Harvey KF (2013) The Hippo size control pathway—ever expanding. Sci Signal 6: pe4. [DOI] [PubMed] [Google Scholar]
- 3. Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ (1995) The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev 9: 534–546. [DOI] [PubMed] [Google Scholar]
- 4. Xu T, Wang W, Zhang S, Stewart RA, Yu W (1995) Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121: 1053–1063. [DOI] [PubMed] [Google Scholar]
- 5. Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber D, et al. (2002) Salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110: 467–478. [DOI] [PubMed] [Google Scholar]
- 6. Kango-Singh M, Nolo R, Tao C, Verstreken P, Hiesinger PR, Bellen HJ, et al. (2002) Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 129: 5719–5730. [DOI] [PubMed] [Google Scholar]
- 7. Wu S, Huang J, Dong J, Pan D (2003) Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114: 445–456. [DOI] [PubMed] [Google Scholar]
- 8. Harvey KF, Pfleger CM, Hariharan IK (2003) The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114: 457–467. [DOI] [PubMed] [Google Scholar]
- 9. Pantalacci S, Tapon N, Léopold P (2003) The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol 5: 921–927. [DOI] [PubMed] [Google Scholar]
- 10. Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G (2003) Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol 5: 914–920. [DOI] [PubMed] [Google Scholar]
- 11. Jia J, Zhang W, Wang B, Trinko R, Jiang J (2003) The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev 17: 2514–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lai ZC (2005) Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 120: 675–685. [DOI] [PubMed] [Google Scholar]
- 13. Huang J, Wu S, Barrera J, Matthews K, Pan D (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 122: 421–434. [DOI] [PubMed] [Google Scholar]
- 14. Dong J (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130: 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oh H, Irvine KD (2008) In vivo regulation of Yorkie phosphorylation and localization. Development 135: 1081–1088. 10.1242/dev.015255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu S, Liu Y, Zheng Y, Dong J, Pan D (2008) The TEAD/TEF family protein scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell 14: 388–398. 10.1016/j.devcel.2008.01.007 [DOI] [PubMed] [Google Scholar]
- 17. Zhao B (2008) TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22: 1962–1971. 10.1101/gad.1664408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thompson BJ, Cohen SM (2006) The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell 126: 767–774. [DOI] [PubMed] [Google Scholar]
- 19. Nolo R, Morrison CM, Tao C, Zhang X, Halder G (2006) The bantammicroRNA is a target of the hippo tumor-suppressor pathway. Curr Biol 16: 1895–1904. [DOI] [PubMed] [Google Scholar]
- 20. Neto-Silva RM, de Beco S, Johnston LA (2010) Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev Cell 19: 507–520. 10.1016/j.devcel.2010.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo C, Tommasi S, Liu L, Yee JK, Dammann R, Pfeiffer GP (2007) RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr Biol 17: 700–705. [DOI] [PubMed] [Google Scholar]
- 22. Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, et al. (2007) RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell 27: 962–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang J, Smolen GA, Haber DA (2008) Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res 68: 2789–2794. 10.1158/0008-5472.CAN-07-6205 [DOI] [PubMed] [Google Scholar]
- 24. Zhao B, Wei X, Li W, Udan RS, Yang Q, Joungmok K, et al. (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oka T, Mazack V, Sudol M (2008) Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J Biol Chem 283: 27534–27546. 10.1074/jbc.M804380200 [DOI] [PubMed] [Google Scholar]
- 26. Moya IM, Halder G (2014) Discovering the Hippo pathway protein-protein interactome. Cell Res 24: 137–138. 10.1038/cr.2014.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Genevet A, Tapon N (2011) The Hippo pathway and apico-basal cell polarity. Biochem J 436: 213–224. 10.1042/BJ20110217 [DOI] [PubMed] [Google Scholar]
- 28. Yu FX (2012) Regulation of the Hippo–YAP pathway by G-protein-coupled receptor signaling. Cell 150: 780–791. 10.1016/j.cell.2012.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D (2010) The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem 285: 37159–37169. 10.1074/jbc.M110.152942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev 24: 72–85. 10.1101/gad.1843810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song MS, Song SJ, Kim SJ, Nakayama K, Nakayama KI, Lim D- S (2008) Skp2 regulates the antiproliferative function of the tumor suppressor RASSF1A via ubiquitin-mediated degradation at the G1-S transition. Oncogene 27: 3176–3185. [DOI] [PubMed] [Google Scholar]
- 32. Suryaraja R, Anitha M, Anbarasu K, Kumari G, Mahalingam S (2013) The E3 ubiquitin ligase Itch regulates tumor suppressor protein RASSF5/NORE1 stability in an acetylation-dependent manner. Cell Death Dis 4: e565 10.1038/cddis.2013.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lignitto L, Arcella A, Sepe M, Rinaldi L, Delle Donne R, Gallo A, et al. (2013) Proteolysis of MOB1 by the ubiquitin ligase praja2 attenuates Hippo signalling and supports glioblastoma growth. Nat Commun 4: 1822 10.1038/ncomms2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salah Z, Melino G, Aqeilan RI (2011) Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res 71: 2010–2020. 10.1158/0008-5472.CAN-10-3516 [DOI] [PubMed] [Google Scholar]
- 35. Yeung B, Ho KC, Yang X (2013) WWP1 E3 ligase targets LATS1 for ubiquitin-mediated degradation in breast cancer cells. PLoS One 8: e61027 10.1371/journal.pone.0061027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao L, Chen Y, Ji M, Dong J (2011) KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J Biol Chem 286: 7788–7796. 10.1074/jbc.M110.173468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scheel H, Hofmann K (2003) A novel interaction motif, SARAH, connects three classes of tumor suppressor. Curr Biol 13: R899–900. [DOI] [PubMed] [Google Scholar]
- 38. Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J (2004) Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem J 381: 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ni L, Li S, Yu J, Min J, Brautigam CA, Tomchick DR, et al. (2013) Structural basis for autoactivation of human Mst2 kinase and its regulation by RASSF5. Structure 21: 1757–1768. 10.1016/j.str.2013.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deng Y, Matsui Y, Zhang Y, Lai ZC (2013) Hippo activation through homodimerization and membrane association for growth inhibition and organ size control. Dev Biol 375: 152–159. 10.1016/j.ydbio.2012.12.017 [DOI] [PubMed] [Google Scholar]
- 41. Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, et al. (2010) The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A 107: 8248–8253. 10.1073/pnas.0912203107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, et al. (2014) A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev 28: 432–437. 10.1101/gad.233676.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee JH, Kim TS, Yang TH, Koo BK, Oh SP, Lee K- P, et al. (2008) A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J 27: 1231–1242. 10.1038/emboj.2008.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D (2013) Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 154: 1342–1355. 10.1016/j.cell.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Callus BA, Verhagen AM, Vaux DL (2006) Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J 273: 4264–4276. [DOI] [PubMed] [Google Scholar]
- 46. Wu D, Wu S (2013) A conserved serine residue regulates the stability of Drosophila Salvador and human WW domain-containing adaptor 45 through proteasomal degradation. Biochem Biophys Res Commun 433: 538–541. 10.1016/j.bbrc.2013.03.023 [DOI] [PubMed] [Google Scholar]
- 47. Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N (2010) Kibra is aregulator of the Salvador/Warts/Hippo signaling network. Dev Cell 18: 300–308. 10.1016/j.devcel.2009.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Polesello C, Huelsmann S, Brown NH, Tapon N (2006) The Drosophila RASSF homolog antagonizes the hippo pathway. Curr Biol 16: 2459–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Port F, Chen HM, Lee T, Bullock SL (2014) Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc Natl Acad Sci U S A 111: E2967–2976. 10.1073/pnas.1405500111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nagy V, Dikic I (2010) Ubiquitin ligase complexes: from substrate selectivity to conjugational specificity. Biol Chem 391: 163–169. 10.1515/BC.2010.021 [DOI] [PubMed] [Google Scholar]
- 51. Park BH, Lee YH (2011) Phosphorylation of SAV1 by mammalian ste20-like kinase promotes cell death. BMB Rep 44: 584–589. [DOI] [PubMed] [Google Scholar]
- 52. Hochrainer K, Mayer H, Baranyi U, Binder B, Lipp J, Kroismayr R (2005) The human HERC family of ubiquitin ligases: novel members, genomic organization, expression profiling, and evolutionary aspects. Genomics 85: 153–164. [DOI] [PubMed] [Google Scholar]
- 53. Stevens TJ, Paoli M (2008) RCC1-like repeat proteins: a pangenomic, structurally diverse new superfamily of beta-propeller domains. Proteins 70: 378–387. [DOI] [PubMed] [Google Scholar]
- 54. Scheffner M, Kumar S (2014) Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta 1843: 61–74. 10.1016/j.bbamcr.2013.03.024 [DOI] [PubMed] [Google Scholar]
- 55. Yue T, Tian A, Jiang J (2012) The cell adhesion molecule echinoid functions as a tumor suppressor and upstream regulator of the Hippo signaling pathway. Dev Cell 22: 255–267. 10.1016/j.devcel.2011.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ho LL, Wei X, Shimizu T, Lai ZC (2010) Mob as tumor suppressor is activated at the cell membrane to control tissue growth and organ size in Drosophila. Dev Biol 337: 274–283. 10.1016/j.ydbio.2009.10.042 [DOI] [PubMed] [Google Scholar]
- 57. Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D (2010) Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev Cell 18: 288–299. 10.1016/j.devcel.2009.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baumgartner R, Poernbacher I, Buser N, Hafen E, Stocker H (2010) The WWdomain protein Kibra acts upstream of Hippo in Drosophila. Dev Cell 18: 309–316. 10.1016/j.devcel.2009.12.013 [DOI] [PubMed] [Google Scholar]
- 59. Wehr MC, Holder MV, Gailite I, Saunders RE, Maile TM, Ciirdaeva E, et al. (2013) Salt-inducible kinases regulate growth through the Hippo signalling pathway in Drosophila. Nat Cell Biol 15: 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rodriguez CI, Stewart CL (2007) Disruption of the ubiquitin ligase HERC4 causes defects in spermatozoon maturation and impaired fertility. Dev Biol 312: 501–508. [DOI] [PubMed] [Google Scholar]
- 61. Zhou H, Shi R, Wei M, Zheng WL, Zhou JY, Ma W- L (2013) The expression and clinical significance of HERC4 in breast cancer. Cancer Cell Int 13: 113 10.1186/1475-2867-13-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flag-tagged Herc4, Su(dx) and Nedd4 were expressed in S2 cells together with Sav-Myc and Ub-HA. After lysis, Sav was analysed for ubiquitylation.
(TIF)
(TIF)
(TIF)
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.