

Abstract
Background and Objective
The oral direct factor Xa inhibitor edoxaban is a P-glycoprotein (P-gp) substrate metabolized via carboxylesterase-1 and cytochrome P450 (CYP) 3A4/5. The effect of rifampin-induced induction of P-gp and CYP3A4/5 on transport and metabolism of edoxaban through the CYP3A4/5 pathway was investigated in a single-dose edoxaban study.
Methods
This was a phase 1, open-label, two-treatment, two-period, single-sequence drug interaction study in healthy adults. All subjects received a single oral 60 mg edoxaban dose in period 1, and 7 days of 600 mg rifampin (2 × 300 mg capsules once daily) with a single oral edoxaban 60 mg dose administered concomitantly on day 7 in period 2. A 6-day washout period separated the treatments. Plasma concentrations of edoxaban and its metabolites M4 and M6 were measured, and limited assessments of pharmacodynamic markers of coagulation were performed.
Results
In total, 34 healthy subjects were enrolled; 32 completed the study. Coadministration of rifampin with edoxaban decreased edoxaban exposure but increased active metabolite exposure. Rifampin increased apparent oral clearance of edoxaban by 33 % and decreased its half-life by 50 %. Anticoagulant effects based on the prothrombin time (PT) and the activated partial thromboplastin time (aPTT) with and without rifampin at early time points were maintained to a greater-than-expected degree than with edoxaban exposure alone, presumably because of an increased contribution from the active metabolites. Edoxaban was well tolerated in this healthy adult population.
Conclusions
Rifampin reduced exposure to edoxaban while increasing exposure to its active metabolites M4 and M6. PT and aPTT at early time points did not change appreciably; however, the data should be interpreted with caution.
Key Points
| Rifampin induces cytochrome P450 (CYP) 3A4/5 and P-glycoprotein. It also inhibits organic anion–transporting polypeptide (OATP) 1B1. Edoxaban is metabolized (< 10 %) by CYP3A4/5 and is a substrate for P-glycoprotein. Edoxaban’s metabolite, M4, is a substrate for OAT1B1. |
| Coadministration of edoxaban with rifampin reduces total exposure to edoxaban and simultaneously increases exposure to its active metabolites formed via CYP3A4/5. |
| Rifampin also increases exposure to the M4 metabolite, which is not formed through the CYP3A4/5 pathway. |
| Edoxaban was well tolerated in healthy adults. |
Introduction
Edoxaban is an oral direct factor Xa inhibitor [1], which is approved in Japan and the USA [2, 3] and is under consideration for approval in the European Union. Oral administration of edoxaban results in peak plasma concentrations within 1–2 h [4]. Edoxaban undergoes limited metabolism through phase 1 and phase 2 enzymes. Phase 1 metabolism is predominantly mediated by carboxylesterase-1 (CES-1), with a lesser contribution of cytochrome P450 (CYP) 3A4/5 [5]. CES-1 mediates formation of human-specific metabolite M4, while CYP3A4/5 mediates formation of M6 [5]. Both are active metabolites with anticoagulant activity similar to that of the parent drug (data on file). However, because of low abundance (<10 % for each) [5], these metabolites are not expected to contribute significantly to total anticoagulant activity in normal circumstances. Edoxaban is a substrate for the efflux transporter P-glycoprotein (P-gp), and M4 is a substrate for uptake transporter organic anion–transporting polypeptide (OATP) 1B1 [6]. Previous drug–drug interaction studies demonstrated increased exposure to edoxaban when it was coadministered with P-gp inhibitors such as quinidine, verapamil and dronedarone (increases of 77, 53 and 85 %, respectively) [7].
Interactions involving transporters may affect drug or metabolite exposure and safety profiles in various tissues. Thus, the US Food and Drug Administration recommends evaluation of the effects of transporter-based drug interactions on absorption and disposition of investigational drugs [8]. Rifampin is a potent inducer of CYP3A4/5 and P-gp [9, 10]. Rifampin is also an inhibitor of OATP1B1 [11], which is involved in the transport of the active metabolite M4 [6]. This drug interaction study evaluated the effect of multiple doses of rifampin on the single-dose pharmacokinetics of edoxaban and its active metabolites M4 and M6. A limited assessment of the anticoagulatory effect was included at 2, 4 and 24 h after edoxaban dosing.
Methods
Study Design and Objectives
This was a phase 1, open-label, two-treatment, two-period, fixed-sequence study conducted at a single centre. The primary objective was to evaluate the effect of multiple doses of rifampin on the single-dose pharmacokinetics of edoxaban when it was coadministered with rifampin. Plasma concentrations of edoxaban and its active metabolites M4 and M6, and the pharmacodynamic markers prothrombin time (PT), international normalized ratio (INR) and activated partial thromboplastin time (aPTT) were measured. This study was nonrandomized because of the potential carryover effects of CYP and P-gp induction by rifampin.
During period 1, all subjects received a single oral 60 mg edoxaban dose on day 1. During period 2, all subjects received once-daily rifampin 600 mg (2 × 300 mg Rifadin® capsules; Sanofi Aventis, Bridgewater, NJ, USA) on days 1 through 7, along with a single oral dose of edoxaban 60 mg administered concomitantly on day 7. There was a 6-day washout between treatment periods. All treatments were administered following an overnight 10-h fast, and food intake was prohibited for an additional 2 or 4 h following administration of rifampin or edoxaban, respectively. Edoxaban 60 mg was selected because it was the primary therapeutic dose used in phase 3 studies [12, 13] and is well tolerated in healthy subjects [4]. A single rifampin 600 mg dose is the highest recommended dose for treatment of tuberculosis [14].
The study was approved by the Celerion (formerly MDS Pharma Services) Institutional Review Board (Lincoln, NE, USA) and conducted in compliance with the Declaration of Helsinki and the International Conference on Harmonisation consolidated Guideline E6 for Good Clinical Practice. All subjects provided written informed consent prior to screening.
Subjects
Healthy males and nonpregnant/nonbreastfeeding females, 18–45 years of age with a body mass index of 18–32 kg/m2, were enrolled. Subjects were excluded if they had taken any prescription or over-the-counter medications within 14 days of the first dose of study medication, or if they had taken St. John’s wort, anticoagulants, coagulants, antiplatelet therapy, nonsteroidal anti-inflammatory drugs or any drug or substance known to be a strong CYP3A4/5 or P-gp inducer or inhibitor within 30 days of the first dose of study medication. Subjects with a history or risk of bleeding or coagulopathy were excluded.
Sample Collection
Blood samples were collected for assessment of plasma concentrations of edoxaban, M4 and M6 on day 1 of period 1 and on day 7 of period 2 at 0 h (before edoxaban dosing) and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 60 and 72 h after edoxaban dosing. Blood samples were collected for assessment of rifampin plasma concentrations measured at trough on days 1 through 7 of period 2 at 0 h (before rifampin dosing) to confirm rifampin exposure. Blood samples for pharmacodynamic analyses (PT and aPTT) were collected on day 1 of period 1 and on day 7 of period 2 at 0 h (before edoxaban dosing) and at 2, 4 and 24 h after edoxaban dosing.
Bioanalytical Methods
Plasma concentrations of edoxaban and its metabolites were analysed by Advion BioServices (Indianapolis, IN, USA) using two validated liquid chromatography–tandem mass spectrometry methods.
For simultaneous measurement of edoxaban and M4, plasma samples (200 μL) were mixed with internal standards (d6-edoxaban and d3-M4) and loaded onto 96-well Oasis MCX™ (mixed-mode ion exchange resin) solid-phase extraction plates (Waters Corporation, Milford, MA, USA). Analysis of extracted samples was performed by gradient chromatography (flow rate 0.3 mL/min) with 5 mM ammonium acetate (pH 7.0) and methanol, using an Agilent Zorbax Eclipse XDB Phenyl (2.1 mm × 50 mm, 5 μm) column (Agilent Technologies, Santa Clara, CA, USA).
For M6 measurement, plasma samples (300 μL) were mixed with internal standards (d6-M6) and pH adjustment solution (2 % formic acid, 200 μL), centrifuged and loaded onto 96-well Waters MCX™ (mixed-mode cation exchange resin) solid-phase extraction blocks (Waters Corporation). Analysis of extracted samples was performed by gradient chromatography (flow rate 0.5 mL/min) with 5 mM ammonium acetate (pH 7.0) and methanol over a Waters Xbridge Phenyl (2.1 mm × 50 mm, 3.5 μm) column (Waters Corporation).
For both methods, eluted analytes and internal standards were detected by an API4000 mass spectrometer (SCIEX, Framingham, MA, USA) equipped with a TurboIonSpray source (SCIEX) in positive ion mode. For M4 and edoxaban, the reaction monitoring transitions were 550.4 to 366.3 (edoxaban), 556.4 to 372.3 (d6-edoxaban), 521.4 to 339.3 (M4) and 524.4 to 342.3 (d3-M4), respectively. For M6, the reaction monitoring transitions were 534.4 to 352.1 (M6) and 540.2 to 358.1 (d6-M6), respectively. The calibration curves for edoxaban (1/x2 weighting, quadratic), M4 (1/x2 weighting, linear) and M6 (1/x weighting, linear) ranged from 0.764 to 382 ng/mL, from 0.0792 to 7.92 ng/mL and from 0.1 to 5 ng/mL, respectively. Samples with plasma concentrations >382 ng/mL were diluted with appropriate controls. In assay validations for edoxaban, the intra- and interassay precision (coefficient of variation [CV]) values were ≤11.0 and ≤8.8 %, respectively, for edoxaban quality control (QC) samples prepared at 0.764, 2.29, 153 and 306 ng/mL, and the intra- and interassay accuracy values were −6.9 to 5.8 %. For M4, the intra- and interassay precision (CV) values were ≤12.3 and ≤11.5 %, respectively, for QC samples prepared at 0.0792, 0.238, 3.17 and 6.34 ng/mL, and the intra- and interassay accuracy values were −2.0 to 18.5 %. For M6, the intra- and interassay precision (CV) values were ≤9.3 and ≤6.7 %, respectively, for QC samples prepared at 0.1, 0.3, 2.0 and 4.0 ng/mL, and the intra- and interassay accuracy values were −2.3 to 16.2 %.
Biomarkers (PT and aPTT) and rifampin plasma concentrations were analysed by Celerion (formally MDS Pharma Services) (Neptune, NJ, USA) using validated methods.
Pharmacokinetic and Pharmacodynamic Analyses
Plasma concentration data for edoxaban and its metabolites were analysed by noncompartmental methods using WinNonlin Professional Version 5.2 (Pharsight Corp., Mountain View, CA, USA). Key parameters included the maximum plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration–time curve from time zero extrapolated to infinity (AUC∞) calculated using the log-linear trapezoidal method, and apparent terminal half-life (t½). Apparent total body clearance (CL/F) was calculated for edoxaban only. Pharmacokinetic parameters were estimated using actual sampling time points. Biomarker data for PT and aPTT were analysed using SAS® Version 9.1.3 (Cary, NC, USA).
Safety
Safety was evaluated throughout the study via physical examinations, vital signs, monitoring of body weight and laboratory tests (haematology, serum chemistry, urinalysis and electrocardiograms). In addition, adverse events (AEs) were monitored and recorded according to the Medical Dictionary for Regulatory Activities version 12.0. The incidence, severity (i.e. mild, moderate or severe), duration and potential relationship to the study medication were captured for the AEs.
Statistical Analysis
Continuous pharmacokinetic and pharmacodynamic variables were summarized by descriptive statistics. Statistical analyses were performed in SAS®. Analysis of variance with treatment as a fixed effect and subject as a random effect was performed on the ln-transformed Cmax and AUC∞ obtained for each treatment, and 90 % confidence intervals (CIs) were calculated for the ratios of the geometric least squares means (GLSMs) between treatments. Absence of an effect of rifampin on edoxaban pharmacokinetics was concluded if the 90 % CIs about the GLSM ratios of Cmax and AUC∞ of edoxaban administered with and without rifampin were contained within the equivalence interval of 80–125 %. The tmax values for edoxaban and its metabolites were analysed using a Hodges–Lehmann estimator of the differences, and mean values and CIs were generated with the Moses method.
Assuming an intra-individual CV for Cmax of 24 % for edoxaban, based on prior studies, and assuming that rifampin had little or no effect (ratio <1.05) on edoxaban pharmacokinetics, a sample size of 28 subjects was required to provide at least 80 % power to determine absence of an effect of rifampin on Cmax and AUC of edoxaban with a CI of 90 % around the 80–125 % no-effect boundaries. A total of 34 subjects were enrolled to ensure that 28 subjects would complete the study.
Results
Thirty-four healthy adults aged 21–44 years were enrolled, and 32 subjects completed the study. Table 1 summarizes the demographic and baseline characteristics of the enrolled subjects. One subject was withdrawn because of an AE, and one subject withdrew consent for participation.
Table 1.
Demographic and baseline characteristics (enrolled population, N = 34)
| Characteristic | Value |
|---|---|
| Male, n (%) | 27 (79.4) |
| Race, n (%) | |
| Black | 24 (70.6) |
| White | 7 (20.6) |
| Asian | 2 (5.9) |
| Other | 1 (2.9) |
| Ethnicity, n (%) | |
| Hispanic/Latino | 3 (8.8) |
| Not Hispanic/Latino | 31 (91.2) |
| Age, years | 30.9 ± 6.4a |
| Body mass index, kg/m2 | 26.1 ± 2.7a |
aMean ± standard deviation
The mean plasma edoxaban concentrations were similar for edoxaban alone and for edoxaban with rifampin up to the time when mean Cmax was reached (Fig. 1a). Plasma edoxaban concentrations declined faster when edoxaban was coadministered with rifampin than when it was administered alone.
Fig. 1.
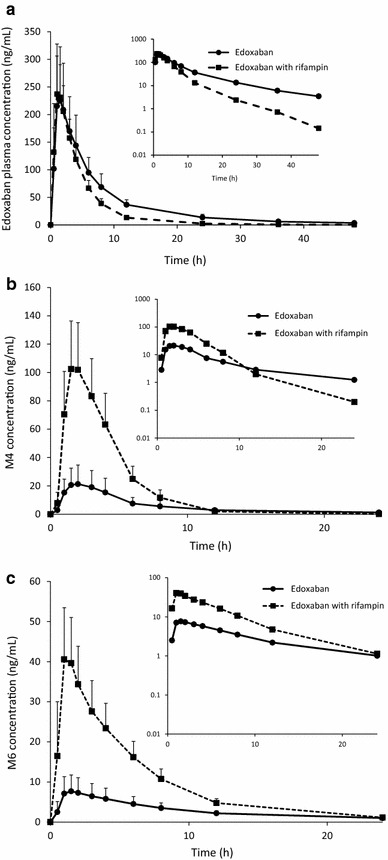
Mean plasma concentration–time curves of edoxaban (a) and the edoxaban metabolites M4 (b) and M6 (c) with and without rifampin. Edoxaban = a single oral dose of edoxaban 60 mg. Edoxaban with rifampin = 7 days of rifampin 600 mg (2 × 300 mg capsules once daily) followed by a single oral dose of edoxaban 60 mg administered concomitantly on day 7. Error bars represent standard deviations. The insets show semi-log scale plots
Edoxaban Cmax and tmax were similar between treatments (Table 2). Concomitant administration of rifampin with edoxaban resulted in an approximate 34 % decrease in total exposure to edoxaban (AUC∞) when compared with administration of edoxaban alone (Table 3). Rifampin coadministration resulted in an approximate 50 % reduction in edoxaban t½ and an approximate 33 % increase in CL/F.
Table 2.
Pharmacokinetic parameters of edoxaban and its metabolites
| Parameter | Edoxaban | M4 | M6 | |||
|---|---|---|---|---|---|---|
| Edoxaban alone, N = 34 | Edoxaban with rifampin, N = 32 | Edoxaban alone, N = 34 | Edoxaban with rifampin, N = 32 | Edoxaban alone, N = 34 | Edoxaban with rifampin, N = 32 | |
| C max, ng/mL | 243 ± 100 | 257 ± 61.8 | 23.1 ± 13.2 | 108 ± 33.0 | 8.55 ± 4.10 | 44.5 ± 12.3 |
| t max, h | 1.08 (0.50–4.00) | 1.00 (0.50–3.00) | 1.81 (1.00–3.05) | 2.00 (1.00–3.00) | 1.49 (0.983–3.00) | 1.00 (0.617–3.00) |
| AUC∞, ng·h/mL | 1835 ± 442 | 1192 ± 214 | 161 ± 69.8 | 449 ± 140 | 91.1 ± 28.2 | 261 ± 57.4 |
| t ½, h | 13.6 ± 6.06 | 6.54 ± 4.24 | 14.3 ± 6.0 | 4.23 ± 3.78 | 14.3 ± 5.58 | 10.0 ± 3.81 |
| CL/F, L/h | 34.8 ± 9.22 | 52.0 ± 9.76 | NC | NC | NC | NC |
| Metabolite to parent exposure ratio (%) | 8.89 ± 3.22 | 38.1 ± 11.8 | 5.09 ± 1.44 | 22.2 ± 4.35 | ||
Data are presented as arithmetic mean ± standard deviation, except for t max values, which are presented as median (range)
AUC ∞ area under the plasma concentration–time curve from time zero extrapolated to infinity, CL/F apparent total body clearance, C max maximum plasma concentration, NC not calculated, t ½ apparent terminal half-life, t max time to C max
Table 3.
Geometric least squares mean ratios for pharmacokinetic parameters
| Parameter | Ratio of edoxaban alone to edoxaban with rifampin (90 % confidence interval) | ||
|---|---|---|---|
| Edoxaban | M4 | M6 | |
| C max | 110.1 (98.8 to 122.6) | 506.4 (446.5 to 574.4) | 551.9 (485.6 to 627.2) |
| AUC∞ | 65.72 (62.5 to 69.1) | 285.5 (264.5 to 308.1) | 291.1 (274.0 to 309.4) |
AUC ∞ area under the plasma concentration–time curve from time zero extrapolated to infinity, C max maximum plasma concentration, t max time to C max
Both M4 and M6 formed rapidly when edoxaban was administered with or without rifampin (Fig. 1b, c). Concurrent edoxaban and rifampin administration increased Cmax values for M4 and M6 by 5- and 4-fold, respectively (Table 2). Coadministration of edoxaban and rifampin produced significant 3-fold increases in metabolite exposure (Table 3). The 90 % CIs for the GLSM ratios were greater than the upper bounds of the 80–125 % intervals for Cmax, AUC∞ and tmax (Table 3). The tmax values for M4 and M6 (Tables 2, 3) were unaffected by coadministration of rifampin with edoxaban; however, the M4 t½ was decreased by approximately 70 % and the M6 t½ was decreased by approximately 35 % when edoxaban was coadministered with rifampin, compared with administration of edoxaban alone (Table 2).
Rifampin plasma concentrations measured at trough each day demonstrated mean (standard deviation [SD]) concentrations of 0.21 (0.48) µg/mL on day 2 and then decreased each day as a consequence of rifampin auto-induction. These values were fairly consistent with historical values for rifampin administered as 600 mg daily for 7 days [15, 16].
The mean baseline PT and aPTT values were similar for both treatments. The PT and aPTT values at 2 and 4 h were minimally different with and without rifampin (Fig. 2). Coadministration of rifampin and edoxaban resulted in minimal changes (<10 %) from the baseline PT and aPTT values, compared with administration of edoxaban alone. However, these data should be interpreted with caution, as only early anticoagulatory effects were assessed.
Fig. 2.
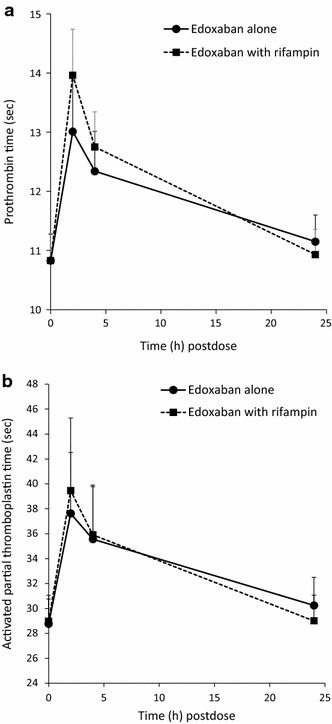
Pharmacodynamic effects of edoxaban with and without rifampin on (a) the mean prothrombin time and (b) the mean activated partial thromboplastin time. Edoxaban = a single oral dose of edoxaban 60 mg. Edoxaban with rifampin = 7 days of rifampin 600 mg (2 × 300 mg capsules once daily) followed by a single oral dose of edoxaban 60 mg administered concomitantly on day 7. The error bars represent standard deviations
Thirty-three subjects (97.1 %) reported 53 AEs during the study. All AEs were mild in intensity. The most common AE was chromaturia (in 32 subjects), a known effect associated with rifampin treatment [14]. The majority of AEs occurred during edoxaban and rifampin coadministration (33 subjects) and were deemed by the investigator to be related to rifampin. Five subjects reported AEs when edoxaban was administered alone. Two AEs were considered related to edoxaban treatment and included headache (reported by one subject) and gingival bleeding (reported by one subject); both events were mild and resolved without treatment. One subject experienced diarrhoea, which was judged to be related to both rifampin and edoxaban. No serious AEs occurred during this study. One subject discontinued participation in the study because of a transient elevation in serum creatinine (21 % above baseline), which was judged by the investigator to be unrelated to the study medication.
Discussion
These results suggest that the effect of rifampin on edoxaban was not only to reduce oral bioavailability and increase excretion through potential induction of P-gp, but also to increase its metabolism to form the metabolite M6 through CYP3A4/5. The increase in M4 is likely due to the inhibitory effect of rifampin on OATP1B1, as well as potential induction of CES-1 through upregulation of the pregnane X receptor gene [16–18]. The exact effects of rifampin on the disposition of the metabolite M4 are not discernible from this study alone and require further studies (e.g. single-dose versus multiple-dose rifampin coadministered with intravenous and oral edoxaban) to better elucidate OATP1B1 inhibitory effects from enzyme/transporter induction effects [18].
The limitations of the study were that the mechanism of the increase in metabolite exposure could not be further elucidated. The administration of edoxaban with a single dose of rifampin may have characterized the effect of rifampin on the inhibition of transporters such as P-gp and OATP1B1. Administration of intravenous edoxaban with and without rifampin may have differentiated the prehepatic versus hepatic effects of rifampin on parent and metabolite exposure. Finally, urine concentrations of edoxaban were not measured. Since renal clearance represents approximately 50 % of the total clearance of edoxaban, the impact of rifampin on transporters in renal clearance were not characterized.
These results demonstrate a drug interaction of edoxaban and its metabolites with rifampin. Induction of both P-gp and CYP3A4/5 by rifampin explains the decreased exposure to edoxaban and the increased exposure to M6 (a metabolite formed via CYP3A4/5). Dosing recommendations in the US prescribing information [3] suggest that concomitant use of edoxaban with rifampin should be avoided because of decreased edoxaban exposure.
Conclusion
Rifampin significantly decreased exposure to edoxaban and increased exposure to its active metabolites.
Acknowledgments
This study was sponsored by Daiichi Sankyo. Writing and editorial support was provided by Terri Schochet, PhD, of AlphaBioCom, LLC, and was funded by Daiichi Sankyo.
Conflict of interest
Jeanne Mendell, Shuquan Chen, Ling He and Madhuri Desai are employees of, and hold stock or stock options in, Daiichi Sankyo Pharma Development. At the time of writing, Dolly A. Parasrampuria was an employee of Daiichi Sankyo.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Footnotes
At the time of writing, Dolly A. Parasramupria was an employee of Daiichi Sankyo.
References
- 1.Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, et al. DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost. 2008;6(9):1542–1549. doi: 10.1111/j.1538-7836.2008.03064.x. [DOI] [PubMed] [Google Scholar]
- 2.Lixiana® (edoxaban tosilate hydrate tablets): full prescribing information. Tokyo: Daiichi Sankyo Company, Ltd.; 2014.
- 3.Savaysa™ (edoxaban) tablets for oral use: full prescribing information. Parsippany: Daiichi Sankyo Inc.; 2015.
- 4.Ogata K, Mendell-Harary J, Tachibana M, Masumoto H, Oguma T, Kojima M, et al. Clinical safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel factor Xa inhibitor edoxaban in healthy volunteers. J Clin Pharmacol. 2010;50(7):743–753. doi: 10.1177/0091270009351883. [DOI] [PubMed] [Google Scholar]
- 5.Bathala MS, Masumoto H, Oguma T, He L, Lowrie C, Mendell J. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos. 2012;40(12):2250–2255. doi: 10.1124/dmd.112.046888. [DOI] [PubMed] [Google Scholar]
- 6.Mikkaichi T, Yoshigae Y, Masumoto H, Imaoka T, Rozehnal V, Fischer T, et al. Edoxaban transport via P-glycoprotein is a key factor for the drug’s disposition. Drug Metab Dispos. 2014;42(4):520–528. doi: 10.1124/dmd.113.054866. [DOI] [PubMed] [Google Scholar]
- 7.Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S, et al. Drug-drug interaction studies of cardiovascular drugs involving P-glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs. 2013;13(5):331–342. doi: 10.1007/s40256-013-0029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Center for Drug Evaluation and Research. Guidance for industry—safety testing of drug metabolites. Rockville: US Food and Drug Administration; 2012. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf.
- 9.Schuetz EG, Schinkel AH, Relling MV, Schuetz JD. P-glycoprotein: a major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc Natl Acad Sci. 1996;93(9):4001–4005. doi: 10.1073/pnas.93.9.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104(2):147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36(1):164–172. doi: 10.1053/jhep.2002.34133. [DOI] [PubMed] [Google Scholar]
- 12.Giugliano RP, Ruff CT, Braunwald E, Murphy SA, Wiviott SD, Halperin JL, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369(22):2093–2104. doi: 10.1056/NEJMoa1310907. [DOI] [PubMed] [Google Scholar]
- 13.Investigators Hokusai VTE, Buller HR, Decousus H, Grosso MA, Mercuri M, Middeldorp S, et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369(15):1406–1415. doi: 10.1056/NEJMoa1306638. [DOI] [PubMed] [Google Scholar]
- 14.Rifadin® rifampin capsules USP: package insert. Bridgewater: Sanofi-Aventis, US, LLC; 2013. Available at http://products.sanofi.us/rifadin/Rifadin.pdf.
- 15.Borin MT, Chambers JH, Carel BJ, Gagnon S, Freimuth WW. Pharmacokinetic study of the interaction between rifampin and delavirdine mesylate. Clin Pharmacol Ther. 1997;61(5):544–553. doi: 10.1016/S0009-9236(97)90134-X. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Cheng X, Aleksunes L, Klaassen CD. Transcription factor-mediated regulation of carboxylesterase enzymes in livers of mice. Drug Metab Dispos. 2012;40(6):1191–1197. doi: 10.1124/dmd.111.043877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Raymond K. Roles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob. 2006;5:3. doi: 10.1186/1476-0711-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam JL, Shugarts SB, Okochi H, Benet LZ. Elucidating the effect of final-day dosing of rifampin in induction studies on hepatic drug disposition and metabolism. J Pharmacol Exp Ther. 2006;319(2):864–870. doi: 10.1124/jpet.106.108282. [DOI] [PubMed] [Google Scholar]