

Abstract
Long QT syndrome (LQTS) is an arrhythmogenic disorder that can lead to sudden death. To date, mutations in 15 LQTS-susceptibility genes have been implicated. However, the genetic cause for approximately 20% of LQTS patients remains elusive. Here, we performed whole-exome sequencing analyses on 59 LQTS and 61 unaffected individuals in 35 families and 138 unrelated LQTS cases, after genetic screening of known LQTS genes. Our systematic analysis of familial cases and subsequent verification by Sanger sequencing identified 92 candidate mutations in 88 genes for 23 of the 35 families (65.7%): these included eleven de novo, five recessive (two homozygous and three compound heterozygous) and seventy-three dominant mutations. Although no novel commonly mutated gene was identified other than known LQTS genes, protein-protein interaction (PPI) network analyses revealed ten new pathogenic candidates that directly or indirectly interact with proteins encoded by known LQTS genes. Furthermore, candidate gene based association studies using an independent set of 138 unrelated LQTS cases and 587 controls identified an additional novel candidate. Together, mutations in these new candidates and known genes explained 37.1% of the LQTS families (13 in 35). Moreover, half of the newly identified candidates directly interact with calmodulin (5 in 11; comparison with all genes; p=0.042). Subsequent variant analysis in the independent set of 138 cases identified 16 variants in the 11 genes, of which 14 were in calmodulin-interacting genes (87.5%). These results suggest an important role of calmodulin and its interacting proteins in the pathogenesis of LQTS.
Introduction
Long QT syndrome (LQTS) is characterized by a prolonged QT interval in the electrocardiogram (ECG) and ventricular tachyarrhythmia. Arrhythmia is often triggered by exercise, particularly swimming, or emotional stress, resulting in recurrent syncope, seizures, and sometimes, sudden, unexpected cardiac death [1].
LQTS has an estimated prevalence as high as one in 2,000 people [2]. To date, mutations in 15 susceptibility genes have been identified. The majority of those affected have mutations in KCNQ1 (LQT1), KCNH2 (LQT2) and SCN5A (LQT3), encoding potassium and sodium ion channel alpha-subunits. These three genes account for 75% of LQTS cases (LQT1: 30%-35%, LQT2: 25%-30%, LQT3: 5%-10%), while the remaining known LQTS genes, which encode beta subunits of plasma membrane channels, channel-interacting proteins, structural membrane scaffolding proteins or membrane anchoring proteins, account for only 5% of cases [3]. Mutations have not been detected in the remaining 20% of patients.
Whole-exome sequencing (WES) is widely used to identify genetic variations in coding regions [4]. WES is more powerful and cost-effective for exonic regions than whole-genome sequencing because it obtains a deeper coverage of the target regions. WES has been recently used to successfully identify causal mutations of Mendelian diseases [5, 6] and driver mutations in tumors [7–9].
Here, we report the identification of candidate pathogenic mutations, through WES and validated by Sanger sequencing, in two-thirds of the examined LQTS families. Although no commonly mutated gene was identified other than known genes, protein-protein interaction (PPI) network analysis revealed that ten candidates interact with proteins encoded by known LQTS genes. Interestingly, half of these directly interact with calmodulin, which is statistically significant when compared to the number of molecules that directly interact with calmodulin. In addition, candidate gene based association studies using an independent set of unrelated LQTS individuals and unaffected individuals identified an additional novel LQTS candidate. Examination of the presence of mutations in these candidate genes in the unrelated LQTS cases revealed that most mutations were in calmodulin-interacting genes. We believe these findings contribute to a greater understanding of LQTS and provide clues for future research into its pathogenic mechanism.
Materials and Methods
Ethics Statement
This study was approved by the ethics committee of the Institutes of National Cerebral and Cardiovascular Center and RIKEN. The design and performance of the current study involving human subjects were clearly described in a research protocol. All participants provided written informed consent before taking part in this research.
Study subjects
LQTS is diagnosed using the following criteria: patients with a Schwartz risk score > 3.5 in the absence of a secondary cause for QT prolongation [10], and/or an unequivocally pathogenic mutation in one of the LQTS genes, or QTc > 500 ms in repeated 12-lead ECG in the absence of a secondary cause for QT prolongation. Among the LQTS patients registered at National Cerebral and Cardiovascular Center who provided written informed consent, we recruited 186 genetically unrelated LQTS cases whose mutations were not detected by genetic screening of known LQTS genes. Among them, 35 had family data (21 family trio data of LQTS patients with unaffected parents and 14 pedigrees with at least one additional LQTS family member, S1 Fig) with DNA samples, and therefore were selected for pedigree analysis. Therefore, among the 35 families, excluding the proband, an additional 85 family members (24 LQTS and 61 unaffected control subjects) were recruited for this analysis. We also included the remaining 151 samples with no family data as genetically independent LQTS cases. In total, 271 samples were subjected to WES analysis. In the course of the analysis, we detected mutations in known LQTS genes for 13 out of 151 non-pedigree cases (Table 1) [11–14] and these individuals were excluded from further analysis. Consequently, we examined 59 LQTS and 61 unaffected individuals in 35 families and 138 unrelated LQTS cases (n = 258). The participant summary, including gender, average age, and the other clinical information, is shown in Table 2.
Table 1. Identification of known-gene and disease-causing variant in the LQTS.
| Phenotype | Gene symbol | Transcript ID | cDNA level change | Protein level change | QTc (ms) | Symptoms | HGMD † , others |
|---|---|---|---|---|---|---|---|
| LQT1* | KCNQ1 | NM_000218.2 | c.760G>A | p.V254M | 570 | syncope | CM960898 [11] |
| LQT1* | KCNQ1 | NM_000218.2 | c.965C>T | p.T322M | 474 | asympt | CM057152 [12] |
| LQT1 | KCNQ1 | NM_000218.2 | c.683+2T>G | - | 470 | Asymp | Pedigree analysis |
| LQT1 | KCNQ1 | NM_000218.2 | c.1032+1G>A | - | 572 | TdP VF Sym40yo | Pedigree analysis |
| LQT2* | KCNH2 | NM_000238.3 | c.1849T>C | p.F617L | 475 | asympt | |
| LQT2* | KCNH2 | NM_000238.3 | c.1831T>G | p.Y611D | 490 | VF | CM107399 [13] |
| LQT2* | KCNH2 | NM_000238.3 | c.307+2T>A | - | 548 | asympt | |
| LQT3* | SCN5A | NM_001160160.1 | c.4900G>A | p.V1634I | 448 | TdP | |
| LQT4* | ANK2 | NM_001148.4 | c.2474C>T | p.T825I | 436 | syncope | |
| LQT4* | ANK2 | NM_001148.4 | c.4876A>G | p.K1626E | 650 | syncope | |
| LQT4* | ANK2 | NM_001148.4 | c.6149T>C | p.I2050T | 464 | VF | |
| LQT4* | ANK2 | NM_001148.4 | c.8123T>C | p.V2708A | 420 | asympt | |
| LQT5 | KCNE1 | NM_000219.3 | c.253G>A | p.D85N | 492 | asympt | CM040436 [14], Pedigree analysis, rare variant |
| LQT9 | CAV3 | NM_033337.2 | c.37A>T | p.I13F | 466 | asympt | Pedigree analysis, SNV |
| LQT11* | AKAP9 | NM_147185.2 | c.2295T>A | p.D765E | 453 | asympt | |
| LQT11* | AKAP9 | NM_147185.2 | c.5341T>A | p.S1781T | 457 | asympt | |
| LQT12* | SNTA1 | NM_003098.2 | c.1498C>T | p.R500C | 444 | asympt | |
| NOS1AP | NM_014697.2 | c.1276G>A | p.V426M | 413 | asympt | disease causing variant | |
| NOS1AP | NM_014697.2 | c.824C>T | p.S275F | 453 | syncope | disease causing variant |
asympt; asymptomatic, SNV: single nucleotide variant
†accession number obtained from HGMD professional (ver. 2014.4, accessed on Mar. 19, 2015)
*mutations detected in known LQTS genes for non-pedigree cases.
Table 2. Clinical background of LQTS patients and their family members.
| 35 LQTS families | unrelated LQTS (n = 138) | ||
|---|---|---|---|
| LQTS (n = 59) | Control (n = 61) | ||
| age | 23±18 | 25±18 | 19±16 |
| male/ female | 20/39 | 36/25 | 54/84 |
| QTc (ms) | 480±40 | 402±21 | 466±49 |
| syncope, n (%) | 24 (41) | 1 (1) | 59 (43) |
| VF or CA, n (%) | 9 (15) | 0 (0) | 18 (13) |
VF: ventricular fibrillation, CA: cardiac arrest
Whole-exome sequencing
Exome capture was performed by the Agilient SureSelect Human All Exon V4 according to the manufacturer’s instructions. This kit captures genomic DNA by in-solution hybridization with RNA oligonucleotides, enabling specific targeting of approximately 51Mb of the human genome. The captured DNA was sequenced using the Illumina HiSeq2000 platform with paired-end reads of 101bp for insert libraries of 150–200bp according to the manufacturer’s instructions.
Exome sequence data analysis
Read sequences were mapped by the Burrows-Wheeler Aligner (BWA: version 0.6.1) [15] to the human reference genome (GRCh37). Duplicate PCR reads were identified and removed using SAMtools (version 0.1.8) [16] and in-house software. After filtering by pair mapping distance, mapping uniqueness and pair orientation, the mapping result files were converted into pileup format using SAMtools. Variant calling was conducted based on methods we have published elsewhere, VCMM [17]. We used the following quality control filters: (i) alignments near putative indels were refined using GATK [18]; (ii) a stand bias filter excluded variants whose alternative allele was preferentially found in one of the two available read orientations at the site.
Variants that were found in dbSNP (version 137) [19], 1000 Genomes Project (n = 1,094) [20], NHLBI Exome Sequencing Project Exome Variant Server (n = 6,503; http://evs.gs.washington.edu/EVS/) [accessed June 2012] (ESP6500) [21] and our in-house whole genome and exome data composed of 1,257 non-cardiac Japanese individuals were excluded from further analyses. Nongenic, intronic and synonymous variants other than those occurring at canonical splice sites and non-synonymous variants predicted as benign/tolerant by both SIFT (http://sift.jcvi.org/www/) [22] and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) [23] were also excluded. Furthermore, we assumed that affected individuals had de novo or recessive (both homozygous and compound heterozygous) mutations for parent/affected offspring trio families and dominant for the other families. All candidate mutations were validated using Sanger sequencing of both the affected and unaffected individuals.
All mutations in known LQTS genes and in candidate genes, identified in this study, have been deposited into NCBI ClinVar with the accession numbers SCV000221974—SCV000222093.
Network analysis
Network analysis was performed using the Ingenuity Pathway Analysis software (IPA; Ingenuity Systems) based on the 15 known LQTS genes and the 88 candidate pathogenic genes identified. We considered molecules and/or relationships available in the IPA Knowledge Base for human, mouse and rat and set the confidence filter to experimentally observed or high (predicted). Networks were generated with a maximum size of 35 genes and allowing up to 10 networks. Molecules in the query set with recorded interactions were eligible for network construction using the IPA algorithm. Networks were ranked by IPA network score according to their degree of relevance to the eligible molecules in the query data set. The network score is calculated using Fisher’s exact test on a basis of the number of eligible molecules in the network and its size, as well as the total number of eligible molecules analyzed and the total number of molecules in the Ingenuity Knowledge Base that could potentially be included in the networks.
Quality control and gene-based association study
We used 748 Japanese individuals, which included 161 LQTS cases (23 probands in LQTS families and 138 independent LQTS patients) and 587 controls. Closely related subjects, where the identity-by-descent (IBD) proportion of alleles shared was over 0.125, and outliers by principal-component analysis (PCA) [24] (S2 Fig) were previously excluded. We estimated the IBD sharing score using PLINK’s ‘-genome’ option [25] and performed PCA using gdsfmt and SNPRelate packages in the statistical software R [26]. We also excluded all SNVs with a genotype call rate < 0.80, a Hardy-Weinberg equilibrium p-value < 1×10-6 or nongenic and intronic variants other than those occurring at canonical splice sites. When also considering a MAF < 0.005, 51,393 SNVs passed these stringent quality control criteria. The quantile-quantile (QQ) plots of the p-values from the Cochran-Armitage test for trend showed the genomic inflation factor λ GC to be 1.027 (S3 Fig).
For the gene-based association studies, we used the SKAT-O test [27], which encompasses both burden tests (e.g. CMC method [28]) and variance-component tests (e.g. SKAT [29]). We performed the analysis using default weights and MAF < 0.01 for the combination of non-synonymous variants predicted to be damaging by SIFT [22] or PolyPhen-2 [23] analysis and splice-site variants. We performed the test for candidate genes with at least two variants and declared a gene-based test association significant when q-value < 0.05.
Results
Identification of candidate mutations in probands
On average, 6.7 Gbp of short read sequence data were obtained from WES (S1 Table). In total, 68.6% of the sequenced bases were mapped to the targeted regions and 92.8% of mapped exon sequences had at least ten times coverage (S4 Fig). The average coverage was 68X across individuals. An average of 19,505 coding SNVs and 516 coding insertion/deletion (indels) were identified per proband with high confidence (S2 Table). We developed an automated pipeline to systematically identify all candidate non-synonymous mutations in each affected individual (Fig 1). We first excluded all synonymous variants other than those occurring at canonical splice sites. This first step reduced the number of candidates to an average of 9,256 non-synonymous and canonical splice site variants per proband. We further reduced this number to 76 variants and 15 coding indels by excluding variants found in public databases; dbSNP137 [19], 1000 Genomes Project [20], NHLBI Exome Variant Server (ESP6500) [21], the Human Genetic Variation Database (HGVD: http://www.genome.med.kyoto-u.ac.jp/SnpDB). We also used our in-house whole exome or whole genome database composed of 1,257 Japanese individuals. We then excluded the variants predicted as benign/tolerant by both SIFT (http://sift.jcvi.org/www/) [22] and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) [23], and finally selected candidate mutations that co-segregated among affected individuals within each of the pedigrees (S2 Table). We identified 92 candidate pathogenic mutations in 88 genes in 23 out of the 35 families (65.7%), all of which were validated by Sanger sequencing. These are eleven de novo, five recessive (two homozygous and three compound heterozygous) and seventy-three dominant mutations (S3 Table). No gene was found to be commonly mutated among pedigrees.
Fig 1. Experimental work flow for detecting sequence variants by WES.
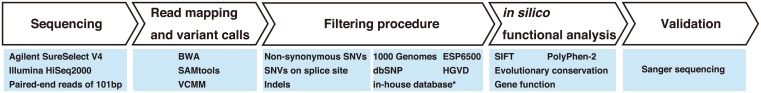
In-house database with asterisk is our in-house whole exome or whole genome data composed of 1,257 non-cardiac Japanese individuals.
Protein-protein interaction (PPI) network analysis
We applied PPI network analysis to a gene set of the 15 known genes and the 88 candidate pathogenic genes identified in this analysis, in order to elucidate any enrichment of functional units or categories. Using Ingenuity Pathways analysis software (IPA; Ingenuity Systems), we identified an interesting network, ranked top in IPA network score, composed of proteins encoded by all 15 known genes and 10 candidate pathogenic genes. Seven of the 10 pathogenic candidates were found to directly interact with at least one protein encoded by known LQTS genes (Fig 2) and contain candidate mutations that occur at evolutionarily conserved amino acids (S5 Fig) which were predicted to be damaging by SIFT [22] or PolyPhen-2 [23] analysis and to have a strong functional impact on the gene (Table 3). In addition, half of the 10 pathogenic candidates were calmodulin-interacting genes (RYR2, UBR4, UBR5, PI4KA and KIF21B) (Fig 2), which was statistically significant when compared to the number of molecules that directly interact with calmodulin (p = 0.042, Fisher’s exact test). We previously reported that calmodulin mutations are associated with LQTS [30]. These results suggest an important role of calmodulin and its interacting proteins in the pathogenesis of LQTS. Through PPI analysis, we could detect candidate mutations in 12 families.
Fig 2. The top-scoring IPA network constructed on the basis of known genes/proteins and candidate pathogenic genes/proteins identified.

The green and pink objects represent known LQTS genes and candidate pathogenic genes identified in this PPI analysis, respectively.
Table 3. Potential pathogenic mutations detected in PPI analysis and Gene based Association Study (GAS) using independent samples.
| ID | Gene | Model† | Transcript ID | cDNA level change | Protein level change | SIFT/PolyPhen-2* | Analysis |
|---|---|---|---|---|---|---|---|
| T02 | WDR26 | De novo | NM_025160.6 | c.612G>T | p.L204F | T/- | PPI |
| T08 | RYR2 | De novo | NM_001035.2 | c.12272C>T | p.A4091V | D/D | PPI |
| T12 | UBR5 | AR (CHTZ) | NM_015902.5 | c.5837A>G | p.H1946R | D/P | PPI |
| c.3752G>A | p.R1251H | D/B | PPI | ||||
| T17 | UBR4 | De novo | NM_020765.2 | c.6397G>A | p.A2133T | T/D | PPI |
| T21 | KIF21B | De novo | NM_017596.2 | c.3601C>T | p.R1201W | D/D | PPI |
| D02 | SLC2A5 | AD | NM_003039.2 | c.808C>T | p.R270W | D/D | GAS |
| D03 | CIT | AD | NM_001206999.1 | c.5786C>A | p.S1929Y | D/D | PPI |
| D04 | KCNQ1 | AD | NM_000218.2 | c.683+2T>G | - | -/- | PPI |
| D07 | CAV3 | AD | NM_033337.2 | c.37A>T | p.I13F | T/B | PPI |
| D08 | KCNQ1 | AD | NM_000218.2 | c.1032+1G>A | - | -/- | PPI |
| D09 | KCNE1 | AD | NM_000219.3 | c.253G>A | p.D85N | D/P | PPI |
| D10 | SIRT6 | AD | NM_016539.2 | c.742C>T | p.R248C | D/D | PPI |
| PIK3CG | AD | NM_002649.2 | c.574G>A | p.D192N | T/D | PPI | |
| D14 | PI4KA | AD | NM_058004.2 | c.247G>A | p.D83N | D/D | PPI |
| RIMS1 | AD | NM_014989.4 | c.1477G>C | p.E493Q | D/D | PPI |
*D = damaging; P = probably damaging; T = tolerated; B = benign.
†AR: autosomal recessive (CHTZ = compound heterozygous), AD: autosomal dominant. Bold: known LQTS genes.
Candidate gene-based association study using an independent set of case/control samples
We could not identify candidate pathogenic genes supported by PPI analysis for the remaining 11 families, although 44 genes were still candidates. Therefore, we performed candidate gene-based association studies using the sequence kernel association optimal test: SKAT-O (Fig 3, see Materials and Methods) [27], in order to identify likely pathogenic genes with cumulative effects in LQTS patients from the 44 candidate genes. We used 11 probands from each of these families, 12 probands from each of the families in which no candidates were identified by pedigree analysis, and a set of 138 genetically unrelated LQTS cases and 587 controls (Fig 3). In total, 161 cases and 587 controls were examined and a significant association in the SLC2A5 gene (also known as GLUT5, FDR-adjusted p-value (q-value) = 0.014, Tables 3 and 4) was found.
Fig 3. Experimental work flow for detecting candidate pathogenic mutations.

Table 4. Significant association of SLC2A5 detected by gene-based association study.
| Case | Control | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Transcript ID | cDNA level change | Protein level change | 11 | 12 | 22 | 11 | 12 | 22 | q-value |
| NM_003039.2 | c.888C>G | p.I296M | 0 | 4 | 157 | 0 | 1 | 585 | 0.014 |
| c.808C>T | p.R270W | 0 | 1 | 160 | 0 | 0 | 587 | ||
| c.457C>G | p.L153V | 0 | 1 | 159 | 0 | 1 | 586 | ||
Candidate pathogenic mutations in an independent set of unrelated LQTS cases
Investigation into the presence of possible mutations in these 11 genes in 138 genetically independent cases identified 16 candidate pathogenic mutations in 15 individuals (Table 5, candidate mutations in both WDR26 and RYR2 were identified in the same individual), which were non-synonymous variants and absent from in-house/public variant databases. Out of the 16 candidate mutations, 14 were calmodulin-interacting genes (87.5%, Fig 2), and 9 of these occurred at evolutionarily conserved amino acids (64.3%): four were missense variants in RYR2, three in UBR4, one in PI4KA and one in KIF21B (Table 5). Functional analysis of these mutations though evolutionarily conserved amino acid residue examination showed these mutations to be strong candidates.
Table 5. Candidate mutations in independent unrelated cases.
| Gene | Transcript ID | cDNA level change | Protein level change | SIFT /PolyPhen-2* | Evolutionally conserved amino acid† |
|---|---|---|---|---|---|
| RYR2 | NM_001035.2 | c.497C>G | p.S166C | D/D | Conserved |
| RYR2 | NM_001035.2 | c.1259G>A | p.R420Q | D/D | |
| RYR2 | NM_001035.2 | c.1298T>C | p.L433P | D/B | Conserved |
| RYR2 | NM_001035.2 | c.5278C>T | p.R1760W | D/D | |
| RYR2 | NM_001035.2 | c.8470C>T | p.R2824W | D/D | |
| RYR2 | NM_001035.2 | c.11017C>T | p.R3673W | D/D | |
| RYR2 | NM_001035.2 | c.12438G>C | p.E4146D | D/D | Conserved |
| RYR2 | NM_001035.2 | c.13780A>C | p.K4594Q | D/D | Conserved |
| UBR4 | NM_020765.2 | c.1097A>G | p.K366R | T/P | Conserved |
| UBR4 | NM_020765.2 | c.1349G>T | p.R450L | D/D | Conserved |
| UBR4 | NM_020765.2 | c.1557G>C | p.Q519H | D/D | Conserved |
| UBR5 | NM_015902.5 | c.2965C>T | p.R989W | -/- | |
| PI4KA | NM_058004.2 | c.738C>G | p.I246M | T/P | Conserved |
| KIF21B | NM_017596.2 | c.2224G>A | p.E742K | D/P | Conserved |
| CIT | NM_001206999.1 | c.5783C>T | p.A1928V | -/- | Conserved |
| WDR26 | NM_001115113.2 | c.59G>A | p.G20E | T/- |
*D = damaging; P = probably damaging; T = tolerated; B = benign.
†Conserved: evolutionally conserved amino acid in seven organisms: Homo sapiens, Macaca mulatta, Mus musculus, Canis familiaris, Gallus gallus, Xenopus tropicalis and Danio rerio.
Only candidate mutations in WDR26 (c.59G>A [p.G20E]) and RYR2 (c.11017C>T [p.R3673W]) were identified in the same individual.
Interestingly, nine (including 6 novel) mutations were identified in the RYR2 gene, which were found in younger patients with no affected family members (Table 6). Many of the patients with the RYR2 mutation had similar exercise-induced cardiac events (4 syncope, 2 VF, 1 cardiac arrest). This frequency was also higher and more severe compared with that in genotype-unknown LQTS, while the QTc interval was shorter in patients with the RYR2 mutation than that with genotype-negative LQTS (439 ± 30 vs. 471 ± 50 ms; p-value = 0.01), strengthening the importance of RYR2 in LQTS pathogenesis.
Table 6. Clinical background of patients with long-QT interval and RYR2 mutation.
| cDNA level change | Protein level change | age | sex | Affected family members | QTc | event |
|---|---|---|---|---|---|---|
| c.497C>G | p.S166C | 11 | F | none | 416 | Syncope during swim, novel |
| c.1259G>A | p.R420Q | 14 | M | none | 412 | Syncope during swim (12 y), SD (17 y) |
| c.1298T>C | p.L433P | 18 | F | none | 452 | VF during exercise (17 y) |
| c.5278C>T | p.R1760W | 16 | M | none | 425 | Syncope during swim, novel |
| c.8470C>T | p.R2824W | 7 | M | none | 439 | Asympt, novel |
| c.11017C>T | p.R3673W | 16 | M | none | 469 | Heart failure, novel |
| c.12272C>T | p.A4091V | 16 | M | none | 443 | CA during exercise |
| c.12438G>C | p.E4146D | 2 | F | none | 401 | VF, novel |
| c.13780A>C | p.K4594Q | 12 | F | none | 496 | Syncope during swim (10 y), novel |
Discussion
We sequenced the exomes of 59 LQTS individuals and 61 unaffected individuals from 35 families and systematically identified candidate mutations in the affected individuals. Subsequent PPI network analysis revealed that a statistically significant proportion of pathogenic candidate molecules interacted directly with calmodulin (RYR2 [31], UBR4 [32], UBR5 [33], PI4KA [34] and KIF21B [34]). Calmodulin is a primary sensor of intracellular calcium levels in eukaryotic cells, playing a key role in the proper mediation of Ca2+ signaling, and interacts with several known LQTS genes (SCN5A [35], SNTA1 [36] and CACNA1C [37]), giving strength to the possibility that these candidate genes also play a pathogenic role in LQTS. In particular, RYR2 has previously been reported as gene associated with several arrhythmic diseases, including LQTS [38], catecholaminergic polymorphic ventricular tachycardia (CPVT) [39–41], arrhythmogenic right ventricular dysplasia type 2 [42–44] and sudden infant death syndrome [45]. Along with one candidate non-synonymous mutation (c.12892G>A [p.V4298M]) in RYR2 that has been previously reported in LQTS [38], we identified nine additional candidate mutations (Table 6), strengthening the importance of RYR2 in LQTS pathogenesis. PPI network analysis also revealed candidate pathogenic genes that interact directly or indirectly with known LQTS genes (RIMS1 [46], CIT [47], PIK3CG [48], SIRT6 [49] and WDR26 [33]), implying that these candidate genes might also cause LQTS. In particular, RIMS1 has been reported to regulate insulin secretory machinery [50]. Since insulin infusion has been shown to cause QTc prolongation in animal models [51, 52], this gene may be more likely to play a pathogenic role in LQTS.
A candidate gene based association study also identified an additional candidate pathogenic gene, SLC2A5, encoding a facilitated glucose/fructose transporter that plays a fundamental role in the pathogenesis of fructose-induced hypertension [53]. Since the mechanistic link between hypertension and fatal arrhythmia is not well-characterized, the role of this gene in the pathogenesis of long QT syndrome requires further investigation.
We examined the presence of mutations in the 11 candidate pathogenic genes in the genetically independent individuals. Most of the mutations were observed in calmodulin-interacting genes or known LQTS interacting genes (15 out of 16, Table 5), and many of these occurred at evolutionarily conserved amino acid across multiple species (10 out of 15). Since amino acid substitutions at evolutionarily conserved positions could potentially lead to deleterious effects on gene functions, these mutations may play an important role in the pathogenesis of LQTS.
To our knowledge, this study is the largest whole-exome sequencing analyses for LQTS. Our analysis revealed several novel candidate pathogenic genes through PPI analysis and gene-based association study. We believe our findings will be an anchor point for finding novel pathogenesis of this disorder.
Supporting Information
Samples with an asterisk were subject to WES analysis and those with a question mark have unknown affected status.
(PDF)
Plot of the first and the second principle components of the 749 subjects along with 45 East Asian (HapMap populations of Japanese in Tokyo: JPT), 45 Han Chinese in Beijing: CHB), 90 African (HapMap population of Yoruba in Ibadan, Nigeria: YRI), and 90 European (HapMap population of Utah, USA residents with ancestry from northern and western Europe: CEU) populations. The one outlier indicated by the arrow (case) was excluded.
(PDF)
The genomic inflation factor λ GC was 1.027.
(TIFF)
Each line corresponds to one of the 120 individuals. On average, 92.8% of all target exons had at least 10-fold coverage.
(PDF)
Homologous sequences were aligned using CLUSTALW. We identified evolutionally conserved amino acid across seven organisms: Homo sapiens, Macaca mulatta, Mus musculus, Canis familiaris, Gallus gallus, Xenopus tropicalis and Danio rerio.
(PDF)
† Proband.
(DOCX)
† NS: non-synonymous SNV, SP: splice-site SNV. * Confirmed candidates: candidates co-segregated in the pedigree and validated using Sanger sequencing.
(DOCX)
† AR: autosomal recessive (HMZ = homozygous, CHTZ = compound heterozygous), AD: autosomal dominant. Bold: LQTS-susceptibility genes.
(DOCX)
Acknowledgments
We thank Keith A Boroevich for critical reading of our manuscript. We also thank the technical staff of the Laboratories for Medical Science Mathematics, Genome Sequencing Analysis and Cardiovascular Diseases at the RIKEN Center for Integrative Medical Sciences for the technical assistance.
Data Availability
Some access restrictions apply to the data underlying the findings. All data relevant for the interpretation of our findings are provided in the paper or the supplementary information, except for the raw sequence data. Genetic data are considered as personal private data in Japan, therefore we are not allowed to submit to a public repository. All mutations in known LQTS genes and in candidate genes, identified in this study, have been deposited into NCBI ClinVar with the accession numbers SCV000221974 - SCV000222093.
Funding Statement
Drs. W. Shimizu, TA, YM, and T. Tanaka were supported in part by the Research Grant for the Cardiovascular Diseases (H24-033, H26-040) from the Ministry of Health, Labour and Welfare, Japan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372(9640):750–63. Epub 2008/09/02. 10.1016/S0140-6736(08)61307-0 S0140-6736(08)61307-0 [pii]. . [DOI] [PubMed] [Google Scholar]
- 2. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761–7. Epub 2009/10/21. 10.1161/CIRCULATIONAHA.109.863209 CIRCULATIONAHA.109.863209 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boczek NJ, Best JM, Tester DJ, Giudicessi JR, Middha S, Evans JM, et al. Exome Sequencing and Systems Biology Converge to Identify Novel Mutations in the L-Type Calcium Channel, CACNA1C, Linked to Autosomal Dominant Long QT Syndrome. Circulation Cardiovascular genetics. 2013. Epub 2013/05/17. doi: CIRCGENETICS.113.000138 [pii]. 10.1161/CIRCGENETICS.113.000138 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, et al. Exome sequencing and the genetic basis of complex traits. Nature genetics. 2012;44(6):623–30. Epub 2012/05/30. 10.1038/ng.2303 ng.2303 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–101. Epub 2009/10/29. 10.1073/pnas.0910672106 0910672106 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nature genetics. 2010;42(1):30–5. Epub 2009/11/17. 10.1038/ng.499 ng.499 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nature genetics. 2011;43(5):442–6. Epub 2011/04/19. 10.1038/ng.810 ng.810 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469(7331):539–42. Epub 2011/01/21. 10.1038/nature09639 nature09639 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–7. Epub 2011/07/30. 10.1126/science.1206923 science.1206923 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124(20):2181–4. 10.1161/CIRCULATIONAHA.111.062182 . [DOI] [PubMed] [Google Scholar]
- 11. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature genetics. 1996;12(1):17–23. 10.1038/ng0196-17 . [DOI] [PubMed] [Google Scholar]
- 12. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. Jama. 2005;294(23):2975–80. 10.1001/jama.294.23.2975 . [DOI] [PubMed] [Google Scholar]
- 13. Itoh H, Shimizu W, Hayashi K, Yamagata K, Sakaguchi T, Ohno S, et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: a Japanese multicenter study. Heart rhythm: the official journal of the Heart Rhythm Society. 2010;7(10):1411–8. 10.1016/j.hrthm.2010.06.013 . [DOI] [PubMed] [Google Scholar]
- 14. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. Journal of the American College of Cardiology. 2009;54(9):812–9. 10.1016/j.jacc.2009.06.005 . [DOI] [PubMed] [Google Scholar]
- 15. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. Epub 2009/05/20. 10.1093/bioinformatics/btp324 btp324 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shigemizu D, Fujimoto A, Akiyama S, Abe T, Nakano K, Boroevich KA, et al. A practical method to detect SNVs and indels from whole genome and exome sequencing data. Sci Rep. 2013;3:2161 Epub 2013/07/09. 10.1038/srep02161 srep02161 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. Epub 2010/07/21. 10.1101/gr.107524.110 gr.107524.110 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000;28(1):352–5. Epub 1999/12/11. doi: gkd114 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Via M, Gignoux C, Burchard EG. The 1000 Genomes Project: new opportunities for research and social challenges. Genome Med. 2010;2(1):3 Epub 2010/03/03. 10.1186/gm124 gm124 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493(7431):216–20. Epub 2012/12/04. 10.1038/nature11690 nature11690 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. Epub 2009/06/30. 10.1038/nprot.2009.86 nprot.2009.86 [pii]. . [DOI] [PubMed] [Google Scholar]
- 23. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. Epub 2010/04/01. 10.1038/nmeth0410-248 nmeth0410-248 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nature genetics. 2008;40(5):646–9. 10.1038/ng.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Team RDC. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2009. [Google Scholar]
- 27. Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13(4):762–75. 10.1093/biostatistics/kxs014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83(3):311–21. 10.1016/j.ajhg.2008.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89(1):82–93. 10.1016/j.ajhg.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Makita N, Yagihara N, Crotti L, Johnson CN, Beckmann BM, Roh MS, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circulation Cardiovascular genetics. 2014;7(4):466–74. 10.1161/CIRCGENETICS.113.000459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hino A, Yano M, Kato T, Fukuda M, Suetomi T, Ono M, et al. Enhanced binding of calmodulin to the ryanodine receptor corrects contractile dysfunction in failing hearts. Cardiovasc Res. 2012;96(3):433–43. Epub 2012/08/16. 10.1093/cvr/cvs271 cvs271 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakatani Y, Konishi H, Vassilev A, Kurooka H, Ishiguro K, Sawada J, et al. p600, a unique protein required for membrane morphogenesis and cell survival. Proc Natl Acad Sci U S A. 2005;102(42):15093–8. Epub 2005/10/11. 0507458102 [pii]. 10.1073/pnas.0507458102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134(5):804–16. Epub 2008/09/09. 10.1016/j.cell.2008.06.048 S0092-8674(08)00835-0 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berggard T, Arrigoni G, Olsson O, Fex M, Linse S, James P. 140 mouse brain proteins identified by Ca2+-calmodulin affinity chromatography and tandem mass spectrometry. J Proteome Res. 2006;5(3):669–87. 10.1021/pr050421l . [DOI] [PubMed] [Google Scholar]
- 35. Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415(6870):442–7. Epub 2002/01/25. 10.1038/415442a 415442a [pii]. . [DOI] [PubMed] [Google Scholar]
- 36. Iwata Y, Pan Y, Yoshida T, Hanada H, Shigekawa M. Alpha1-syntrophin has distinct binding sites for actin and calmodulin. FEBS Lett. 1998;423(2):173–7. Epub 1998/03/25. doi: S0014-5793(98)00085-4 [pii]. . [DOI] [PubMed] [Google Scholar]
- 37. Xiong L, Kleerekoper QK, He R, Putkey JA, Hamilton SL. Sites on calmodulin that interact with the C-terminal tail of Cav1.2 channel. J Biol Chem. 2005;280(8):7070–9. Epub 2004/12/08. doi: M410558200 [pii]. 10.1074/jbc.M410558200 . [DOI] [PubMed] [Google Scholar]
- 38. Kauferstein S, Kiehne N, Erkapic D, Schmidt J, Hamm CW, Bratzke H, et al. A novel mutation in the cardiac ryanodine receptor gene (RyR2) in a patient with an unequivocal LQTS. Int J Cardiol. 2011;146(2):249–50. Epub 2010/12/04. 10.1016/j.ijcard.2010.10.062 S0167-5273(10)00910-1 [pii]. . [DOI] [PubMed] [Google Scholar]
- 39. Hayashi M, Denjoy I, Extramiana F, Maltret A, Buisson NR, Lupoglazoff JM, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119(18):2426–34. Epub 2009/04/29. 10.1161/CIRCULATIONAHA.108.829267 CIRCULATIONAHA.108.829267 [pii]. . [DOI] [PubMed] [Google Scholar]
- 40. Jiang D, Jones PP, Davis DR, Gow R, Green MS, Birnie DH, et al. Characterization of a novel mutation in the cardiac ryanodine receptor that results in catecholaminergic polymorphic ventricular tachycardia. Channels (Austin). 2010;4(4):302–10. Epub 2010/08/03. doi: 12666 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meli AC, Refaat MM, Dura M, Reiken S, Wronska A, Wojciak J, et al. A novel ryanodine receptor mutation linked to sudden death increases sensitivity to cytosolic calcium. Circ Res. 2011;109(3):281–90. Epub 2011/06/11. 10.1161/CIRCRESAHA.111.244970 CIRCRESAHA.111.244970 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001;10(3):189–94. Epub 2001/02/13. . [DOI] [PubMed] [Google Scholar]
- 43. Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97(11):1173–81. Epub 2005/10/22. doi: 01.RES.0000192146.85173.4b [pii]. 10.1161/01.RES.0000192146.85173.4b . [DOI] [PubMed] [Google Scholar]
- 44. Tang Y, Tian X, Wang R, Fill M, Chen SR. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ Res. 2012;110(7):968–77. Epub 2012/03/01. 10.1161/CIRCRESAHA.111.256560 CIRCRESAHA.111.256560 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tester DJ, Dura M, Carturan E, Reiken S, Wronska A, Marks AR, et al. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart rhythm: the official journal of the Heart Rhythm Society. 2007;4(6):733–9. Epub 2007/06/09. doi: S1547-5271(07)00227-5 [pii]. 10.1016/j.hrthm.2007.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coppola T, Magnin-Luthi S, Perret-Menoud V, Gattesco S, Schiavo G, Regazzi R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J Biol Chem. 2001;276(35):32756–62. 10.1074/jbc.M100929200 . [DOI] [PubMed] [Google Scholar]
- 47. Carter CJ. eIF2B and oligodendrocyte survival: where nature and nurture meet in bipolar disorder and schizophrenia? Schizophr Bull. 2007;33(6):1343–53. 10.1093/schbul/sbm007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Viard P, Butcher AJ, Halet G, Davies A, Nurnberg B, Heblich F, et al. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nature neuroscience. 2004;7(9):939–46. 10.1038/nn1300 . [DOI] [PubMed] [Google Scholar]
- 49. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nature medicine. 2012;18(11):1643–50. 10.1038/nm.2961 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gandini MA, Sandoval A, Gonzalez-Ramirez R, Mori Y, de Waard M, Felix R. Functional coupling of Rab3-interacting molecule 1 (RIM1) and L-type Ca2+ channels in insulin release. J Biol Chem. 2011;286(18):15757–65. 10.1074/jbc.M110.187757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Noord C, Sturkenboom MC, Straus SM, Hofman A, Kors JA, Witteman JC, et al. Serum glucose and insulin are associated with QTc and RR intervals in nondiabetic elderly. Eur J Endocrinol. 2010;162(2):241–8. 10.1530/EJE-09-0878 . [DOI] [PubMed] [Google Scholar]
- 52. Drimba L, Dobronte R, Hegedus C, Sari R, Di Y, Nemeth J, et al. The role of acute hyperinsulinemia in the development of cardiac arrhythmias. Naunyn Schmiedebergs Arch Pharmacol. 2013;386(5):435–44. 10.1007/s00210-013-0845-4 . [DOI] [PubMed] [Google Scholar]
- 53. Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem. 2009;284(8):5056–66. 10.1074/jbc.M808128200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Samples with an asterisk were subject to WES analysis and those with a question mark have unknown affected status.
(PDF)
Plot of the first and the second principle components of the 749 subjects along with 45 East Asian (HapMap populations of Japanese in Tokyo: JPT), 45 Han Chinese in Beijing: CHB), 90 African (HapMap population of Yoruba in Ibadan, Nigeria: YRI), and 90 European (HapMap population of Utah, USA residents with ancestry from northern and western Europe: CEU) populations. The one outlier indicated by the arrow (case) was excluded.
(PDF)
The genomic inflation factor λ GC was 1.027.
(TIFF)
Each line corresponds to one of the 120 individuals. On average, 92.8% of all target exons had at least 10-fold coverage.
(PDF)
Homologous sequences were aligned using CLUSTALW. We identified evolutionally conserved amino acid across seven organisms: Homo sapiens, Macaca mulatta, Mus musculus, Canis familiaris, Gallus gallus, Xenopus tropicalis and Danio rerio.
(PDF)
† Proband.
(DOCX)
† NS: non-synonymous SNV, SP: splice-site SNV. * Confirmed candidates: candidates co-segregated in the pedigree and validated using Sanger sequencing.
(DOCX)
† AR: autosomal recessive (HMZ = homozygous, CHTZ = compound heterozygous), AD: autosomal dominant. Bold: LQTS-susceptibility genes.
(DOCX)
Data Availability Statement
Some access restrictions apply to the data underlying the findings. All data relevant for the interpretation of our findings are provided in the paper or the supplementary information, except for the raw sequence data. Genetic data are considered as personal private data in Japan, therefore we are not allowed to submit to a public repository. All mutations in known LQTS genes and in candidate genes, identified in this study, have been deposited into NCBI ClinVar with the accession numbers SCV000221974 - SCV000222093.