

Summary
Heat Shock Transcription Factor 1 (HSF1) is an evolutionarily conserved transcription factor that protects cells from protein misfolding-induced stress and apoptosis. The mechanisms by which cytosolic protein misfolding leads to HSF1 activation have not been elucidated. Here we demonstrate that HSF1 is directly regulated by TRiC/CCT, a central ATP-dependent chaperonin complex that folds cytosolic proteins. A small molecule activator of HSF1, HSF1A, protects cells from stress-induced apoptosis, binds TRiC subunits in vivo and in vitro and inhibits TRiC activity without perturbation of ATP hydrolysis. Genetic inactivation or depletion of the TRiC complex results in human HSF1 activation and HSF1A inhibits the direct interaction between purified TRiC and HSF1 in vitro. These results demonstrate a direct regulatory interaction between the cytosolic chaperone machine and a critical transcription factor that protects cells from proteo-toxicity, providing a mechanistic basis for signaling perturbations in protein folding to a stress-protective transcription factor.
Graphical Abstract
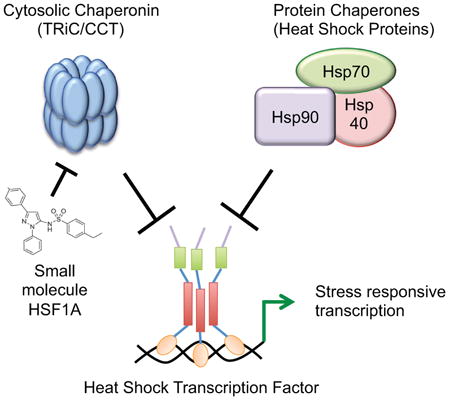
Introduction
Protein misfolding is a biochemical hallmark of diseases that include Alzheimer’s, Parkinson’s and Huntington’s, cardiomyopathy, cataract formation, lysosomal storage disease, Cystic Fibrosis, sickle cell disease and diabetes (Chiti and Dobson, 2006). Specific protein quality control mechanisms operate to both sense and respond to protein misfolding in the endoplasmic reticulum, nucleus, mitochondria and cytosol, resulting in increased folding capacity or degradation of irreversibly damaged proteins. Heat Shock Transcription Factor 1 (HSF1) is a eukaryotic transcription factor that protects cells from cytoplasmic proteo-toxicity and stress-induced apoptosis and is a promising target for neurodegenerative disease therapy (Akerfelt et al., 2010; Fujimoto et al., 2005; Neef et al., 2011). HSF1 serves as a primary mediator of cellular stress responses, facilitating the expression of genes encoding proteins involved in protecting the proteome from stress, including proteins that function in protein folding and degradation as well as transcription, transport, signal transduction, metabolism and a broad array of other adaptive and survival functions in yeast, somatic cells and neurons (Gonsalves et al., 2011; Hahn et al., 2004; Trinklein et al., 2004).
HSF1 is activated in response to a diverse set of environmental conditions associated with cytoplasmic protein misfolding including elevated temperatures, oxidant exposure, metals and bacterial and viral infection. Under normal cell growth conditions HSF1 is largely present as an inactive monomer, where it is thought to be bound and repressed by Hsp40, Hsp70 and Hsp90, abundant protein chaperones that are also involved in the folding and maturation of many cellular proteins including hormone receptors and protein kinases (Shi et al., 1998; Zou et al., 1998). In response to proteo-toxic stress, HSF1 assembles as a homo-trimer, accumulates in the nucleus and binds cis-elements, termed Heat Shock Elements (HSEs) in the promoters of target genes (Akerfelt et al., 2010). HSF1 is post- translationally modified by phosphorylation, sumoylation, ubiquitinylation and acetylation reactions that are proposed to either activate or repress HSF1 function during the regulatory cycle (Cotto et al., 1996; Hietakangas et al., 2003; Sarge et al., 1993; Westerheide et al., 2009). Both HSF1 and Hsp70 possess redox-regulated thiols that also allow intrinsic HSF1 stress-sensing and stress-sensing by repressive protein chaperones, respectively (Ahn and Thiele, 2003; Miyata et al., 2012; Wang et al., 2012). However, the mechanism by which cytoplasmic proteo-toxicity is sensed and transmitted to HSF1 is not well understood.
Sigma 32 (σ32) is a bacterial proteotoxic stress-responsive transcription initiation factor that directs RNA Polymerase to the promoters of protein chaperone genes and other stress-protective target genes. σ32 is regulated by feedback control via direct binding of the DnaK, DnaJ and GroE/L protein folding machinery, functionally analogous to the Hsp70 and Hsp40 chaperones and the TRiC/CCT chaperonin complex of eukaryotic cells, respectively (Guisbert et al., 2004; Rodriguez et al., 2008). These chaperone interactions provide a direct mechanism for the protein folding apparatus to sense and integrate aberrant cellular protein folding with stress-protective responses by modulating σ32 activity and abundance. Here we demonstrate that a chemical activator of HSF1, HSF1A, directly binds the TRiC/CCT chaperonin and modulates TRiC-dependent protein folding. Furthermore, TRiC directly interacts with HSF1 in vitro and represses HSF1-dependent gene activation in vivo. HSF1A antagonizes the repressive HSF1-TRiC interaction, promoting the expression of protein chaperones and other HSF1 target genes that protect cells from protein-misfolding and stress-induced apoptosis. This work establishes a direct regulatory connection between the cytoplasmic protein folding nanomachine TRiC/CCT, and HSF1, a critical transcription factor activated in response to cytoplasmic proteotoxicity.
Results
HSF1A protects against ER stress-induced apoptosis
Previous studies using a humanized HSF1-based yeast screen identified HSF1A, a benzyl-pyrazole-based small molecule, as an activator of human HSF1 (Neef et al., 2010). HSF1A activates HSF1-dependent target gene expression in mammalian and Drosophila cells and ameliorates protein aggregation-mediated toxicity and cell death in neuronal precursor cells and Drosophila models of poly- glutamine (polyQ) mediated protein-misfolding disease (Neef et al., 2010). Since HSF1 activates transcription of target genes that prevent stress-induced apoptosis such as Bag-3, which binds and stabilizes the anti-apoptotic factor Mcl1 (Boiani et al., 2013), HSF1A was evaluated for protection of cells from apoptosis induced by other cell stressors. The endoplasmic reticulum (ER) stress agent, tunicamycin causes the accumulation of misfolded proteins in the ER and promotes activation of the Unfolded Protein Response (UPR), which, upon prolonged activation, induces apoptotic cell death (Glover-Cutter et al., 2013; Walter and Ron, 2011). The proline analogue azetidine (AZC) incorporates into nascent polypeptide chains and promotes their misfolding, causing widespread misfolding of both ER and cytoplasmic proteins. Pre-treatment of cells with HSF1A, followed by tunicamycin or AZC exposure, ameliorated stress-induced cell death (Figure 1 and Figure S1A). Furthermore, HSF1A treatment reduced tunicamycin-induced expression of Bip and Erdj3, two UPR-dependent ER chaperone genes, and blunted activation of caspase-3, the primary mediator of apoptotic cell death (Walter and Ron, 2011) (Figure 1B). The observed protection from apoptotic cell death upon treatment with HSF1A is dependent on the HSF1 (Figure S1B) and HSF1 targets, Hsp70 and Bag-3, have been shown to ameliorate ER-stress induced apoptosis by enhancing UPR signaling and stabilizing anti-apoptotic proteins, respectively (Gupta et al., 2010; Boiani et al., 2013). As ER-stress induced apoptosis contributes to the pathology of diabetes and cardiovascular disease (Scheuner and Kaufman, 2008; Zhang et al., 2012; Zhou et al., 2005), we tested whether HSF1A protects cells in disease-relevant models of ER-stress. Previous studies showed that exposure of the pancreatic beta-cell line INS 832/13 to free fatty acids or high levels of glucose promotes UPR activation and activates apoptosis (El-Assaad et al., 2003). While palmitate caused a marked reduction in INS 832/13 cell viability (Figure 1C) and promoted the expression of the UPR and caspases-3 activation (Figure 1D) both phenotypes were ameliorated by HSF1A pretreatment, as was apoptosis induced by prolonged exposure to high levels of glucose (Figure S1C). Elevated levels of homocysteine induce inflammatory toxicity and is a risk factor in cardiovascular disease, particularly in endothelial cells which fail to express cystathionine β-synthase (CBS) (Wang et al., 1992; Zhang et al., 2001). Since mouse NIH3T3 cells are also deficient in the expression of CBS (Skovby et al., 1984) homocysteine exposure results in a reduction in both NIH3T3 cell viability and basal Hsp70 levels, which promotes activation of the UPR and caspase-3 cleavage (Figure 1E, F), phenotypes that were reduced by HSF1A administration. Taken together, the HSF1 activator HSF1A protects cells from a range of disease relevant proteotoxic conditions that induce apoptosis, supporting further investigation into the mechanism by which HSF1A activates HSF1.
Figure 1. HSF1A protects cells from stress-induced apoptosis.

(A) NIH3T3 cells were pretreated with 50 μM HSF1A for 15 h followed by 0.4 ug/ml tunicamycin for 15 h. (B) Total protein was isolated from NIH3T3 cells treated as in (A) and analyzed for Hsp70, Bip, Erdj3 expression and caspase-3 cleavage, with GAPDH as loading control, by immunoblotting. (C) INS 832/13 cells were pretreated with 20 μM HSF1A for 15 h, before the addition of 0.5 mM palmitate/BSA, or BSA alone, followed by a 15 h incubation. (D) Total protein was isolated from INS 832/13 cells treated as in (C) and analyzed as in (B). (E) NIH3T3 cells were pretreated with 50 μM HSF1A for 15 h, before the addition of 5 mM homocysteine followed by a 15 h incubation. (F) Total protein was isolated from NIH3T3 cells treated as in (B). Cell viability was analyzed with Cell Titer Glo. See also Figure S1.
HSF1A directly interacts with TRiC/CCT subunits
Pull-down experiments using a biotinylated form of HSF1A (HSF1A-Biotin) resulted in the enrichment of all subunits of the TRiC/CCT chaperonin in lysates from both yeast and mammalian cells (Neef et al., 2010). As several protein chaperones including Hsp/Hsc70 and Hsp90 have been linked to the regulation of HSF1, experiments were carried out to determine if HSF1A-Biotin associates with these protein chaperones, or with HSF1. While HSF1A-Biotin associated with the TRiC complex as shown by immunoblotting for two TRiC subunits, Tcp1 and Cct3, it did not bind Hsp90, Hsp70, Hsc70, Hsp27, nor did it associate with HSF1 itself (Figure 2A). To ascertain if HSF1A directly binds TRiC, HSF1A-Biotin was incubated with purified bovine TRiC and purified recombinant Hsp70 and captured proteins analyzed by immunoblotting (Figure 2B). Using antibodies against TRiC subunits Cct2 and Cct3 demonstrates that HSF1A directly interacts with TRiC but not with Hsp70. The TRiC complex is composed of eight independently expressed protein subunits that assemble into a dual-ringed hetero-oligomeric structure (Leitner et al., 2012). While HSF1A-Biotin directly binds to TRiC, these experiments do not distinguish whether HSF1A-Biotin binds to individual TRiC subunits or only interacts with the assembled TRiC complex. As shown in Figure 2C, HSF1A-Biotin binds to the Tcp1, Cct2, Cct5 and Cct8 subunits of yeast TRiC (Reissmann et al., 2012) when independently expressed in E. coli. Moreover, HSF1A-Biotin, but not the Hsp90-specific inhibitor Geldanamycin-Biotin, bound an affinity purified GST-Tcp1 subunit that was expressed by in vitro translation (Figure 2D).
Figure 2. HSF1A directly binds the TRiC complex.

(A) HeLa cell extracts were incubated with 100 μM HSF1A-Biotin, proteins purified with neutravidin-agarose, resolved by SDS-PAGE and analyzed by immunoblotting for HSF1, protein chaperones and TRiC subunits. (I, input; B, Biotin; HB, HSF1A-Biotin) (B) Purified bovine TRiC and recombinant human Hsp70 were incubated with 100 μM HSF1A-Biotin, interacting proteins analyzed as in (A). (C) E. coli extracts expressing the indicated yeast TRiC subunits Tcp1, Cct2, Cct5 or Cct8 fused with FLAG tag were incubated with neutravidin-agarose beads only (B, bead) or with 100 μM HSF1A-Biotin (HB) and immunoblotted. (D) GST-Tcp1 was incubated with either DMSO (B), 100 μM HSF1A-Biotin (HB), or 10 μM Geldanamycin-Biotin (GB) and purified with neutravidin-agarose. As a control, GST-Tcp1 was also purified using glutathione-coated agarose beads (GSH). Purified protein was resolved by SDS-PAGE and analyzed by immunoblotting using Tcp1 antibody. (E) Purified bovine TRiC was incubated with DMSO or HSF1A-Biotin or Biotin and TRiC melting was analyzed by thermal denaturation profiling in the presence of SYPRO orange. (F) Fluorescence anisotropy was used to assess the affinity of HSF1A-FITC for purified recombinant Tcp1. Increased fluorescent polarization (mP) indicates binding to HSF1A-FITC in the presence of increasing concentrations of Tcp1 (G) His6-Tcp1 (See also Figure S2C) was incubated with 0.5 μM Biotin or HSF1A-Biotin (HSF1A-B) and captured with neutravidin-agarose beads and immunoblotted.
A direct interaction between HSF1A and TRiC is further supported by the observation that the thermal stability of purified bovine TRiC is reduced in a dose-dependent manner in the presence of HSF1A-Biotin, but not Biotin (Figure 2E and Figure S2A, B). Moreover, fluorescence anisotropy experiments using FITC coupled to HSF1A demonstrated that HSF1A-FITC bound to a purified Tcp1 subunit of TRiC with an affinity of approximately 600 nM. This was validated qualitatively via titration of purified Tcp1 into binding reactions containing 500 nM Biotin or HSF1A-Biotin (Figures 2F, G and Figure S2C, D). Taken together, these data demonstrate that HSF1A associates with TRiC in vivo and in vitro and can engage in interactions with individual TRiC subunits. These results suggest that HSF1A stimulation of HSF1 activity is mediated through the modulation of TRiC upon direct binding.
The ability of HSF1A to modulate TRiC-dependent protein folding activity was assessed by monitoring TRiC-mediated refolding of denatured 35S-labeled Actin in vitro (Thulasiraman et al., 2000a, b). Addition of 200 μM HSF1A reduced TRiC-mediated actin folding by approximately 50% (Figure 3A), while only mildly inhibiting TRiC-dependent ATP-hydrolysis (Figure 3B). Consistent with the observation that HSF1A is not a potent inhibitor of ATP hydrolysis, ATP, but not HSF1A eluted a human Cct4-GFP fusion protein pre-bound to a gamma phosphate-linked ATP-sepharose resin (Duncan et al., 2012)(Figures 4A–D). As shown by example in Figure 4E for Tcp1, all eight distinct TRiC subunits are composed of two equatorial domains that form the ATP binding domain, two hinge regions and a central apical domain that binds substrates. While full-length purified Tcp1 is bound by HSF1A-Biotin, a Tcp1 fragment (designated D3) containing only the second hinge and equatorial domain (B) is sufficient for HSF1A-Biotin binding (Figure 4F, G). Moreover, the presence of the second hinge region is important for HSF1A-Biotin binding, and mutation of three amino acids within this hinge (LDE to AAA), within the context of the full D3 fragment, abrogated HSF1A-Biotin binding (Figure 4H, I). These results demonstrate that HSF1A binds to TRiC and perturbs its folding activity, but that this interaction does not require the bipartite ATP binding pocket on the TRiC Tcp1 subunit.
Figure 3. HSF1A inhibits TRiC in vitro.

(A) Purified TRiC was treated with DMSO or HSF1A and used in actin refolding assays. Folded actin was captured using a DNaseI-agarose resin, resolved by SDS-PAGE and analyzed and quantitated by autoradiography. (B) Relative ATP hydrolysis (normalized ratio ADP/ATP) by purified bovine TRiC in the presence of DMSO or 200 uM HSF1A over time (min).
Figure 4. HSF1A binding to Cct4 requires a hinge region.

(A) Total cell lysate from HEK293T cells expressing human Cct4-GFP was incubated with ATP-sepharose, bound proteins eluted with ATP and eluate analyzed for Cct4 and GFP by immunobloting. (B) Total cell lysate from HEK293T cells expressing an empty vector, GFP, Cct4 or Cct4-GFP were incubated with ATP-sepharose and captured proteins eluted with ATP. GFP fluorescence was measured. (C) Total cell lysate from HEK293T cells expressing Cct4-GFP was incubated with ATP-sepharose as in (A) and washed with Biotin, HSF1A-Biotin or ATP. Eluted proteins were analyzed for Cct4-GFP by immunoblotting with anti-GFP antibody. (D) Protein elutions from (C) were measured for GFP fluorescence. (E) Tcp1 protein map (Kalisman et al., 2013) representing the three fragments D1 (1–152 aa), D2 (153–379 aa) and D3 (380–559 aa) used to analyze Tcp1-HSF1A interactions. (F) E. coli extract expressing yeast Tcp1 fused with a His6 tag was incubated with neutravidin-agarose beads only (beads), 100 μM Biotin or HSF1A-Biotin (HSF1A-B) and immunoblotted with anti-His tag antibody. (G) E. coli extracts expressing yeast Tcp1 fragments D1, D2 or D3 fused to a His6 tag were incubated with neutravidin-agarose beads only (beads), 100 μM Biotin or HSF1A-Biotin (HSF1A-B) and immunoblotted with anti-His tag antibody. (H) E. coli extracts expressing yeast Tcp1-D3 (380–559 aa) or the truncations Tcp1-D3-Tr1 (397–559 aa) and Tcp1-D3-Tr2 (404–559 aa) were incubated with 100 μM Biotin or HSF1A-Biotin (HSF1A-B), purified with neutravidin-agarose beads and immunoblotted with anti-His tag antibody. (I) E. coli extracts expressing the yeast Tcp1-D3 fragment with the triple point mutations LDE395:397AAA, DSL404:406AAA or GGG421:423AAA fused with a FLAG tag were incubated with 100 μM Biotin or HSF1A-Biotin (HSF1A-B), purified with neutravidin-agarose beads and immunoblotted with anti-FLAG antibody.
Compromising TRiC function activates human HSF1 in yeast and mammalian cells
TRiC is essential for S. cerevisiae and mammalian cell viability (Spiess et al., 2004). As HSF1A-Biotin interacts with both yeast and mammalian TRiC, and modulates mammalian TRiC activity in vitro, experiments were conducted to assess whether HSF1A modulates TRiC activity in vivo. Yeast DAmP strains, in which disruption of the TCP1 and CCT8 3′ UTR destabilizes their corresponding mRNA (Breslow et al., 2008), were exposed to HSF1A or DMSO. Low concentrations of HSF1A (10 μM) did not affect the growth rate of a wild-type yeast strain at 30 °C, but reduced the growth rate of a tcp1-DAmP strain by ~50% (Figure 5A), a phenotype that was exacerbated when the tcp1-DAmP strain was grown under a mild thermal stress of 37 °C. While 10 μM HSF1A did not inhibit growth of the cct8-DAmP strain at 30 °C, growth was significantly reduced at 37 °C. Collectively, these data suggest that HSF1A inhibits yeast TRiC function in vivo.
Figure 5. HSF1A inhibits TRiC function in vivo.

(A) Wild-type, tcp1-DAmP and cct8-DAmP S. cerevisiae strains were treated with DMSO or 10 μM HSF1A, grown at 30 °C or 37 °C for 10 h and growth monitored by optical density. Data shown are growth rate of HSF1A treated cultures as a function of DMSO treated cultures. (B) NIH3T3 cells were treated with DMSO or HSF1A for 15 h or heat shocked for 2 h at 42 °C followed by a 15 h recovery at 37 °C. Total protein was analyzed for Hsp70, HSF1, Cct2, Cct3, α-tubulin, actin and VHL expression by immunoblotting. (C) HeLa cells transfected with a plasmid expressing HA-VHL were treated with DMSO or 80 μM HSF1A for 1 h. HA-VHL and interacting proteins were captured using an anti-HA agarose resin and analyzed by immunobloting for Hsp70, Cct3, Cct8 and VHL. (D) Immunoprecipitated (IP) protein and input (in) protein levels from (C) were quantified and IP protein levels normalized using input protein levels. Data are shown as a percent of IP protein levels of HSF1A treated cells versus DMSO treated cells.
Hsp90 is required for the folding and stability of a number of client proteins and genetic or pharmacological inhibition of Hsp90 promotes the degradation of client proteins (Picard et al., 1990; Schulte et al., 1997). While cells exposed to HSF1A exhibited increased Hsp70 levels as expected due to HSF1 activation, the steady state levels of actin and α-tubulin, two TRiC-client proteins, were not altered (Figure 5B, Figure S3). A modest reduction in von-Hippel-Lindau tumor suppressor protein (VHL) levels was observed in response to HSF1A, though this reduction was dramatic in response to heat shock and may result from cell stress rather than inhibition of TRiC activity. Modest increases in Cct2 and Cct3 levels were also observed in response to HSF1A (Figure 5B, Figure S3), consistent with the mammalian TRiC genes being direct HSF1 targets (Kubota et al., 1999).
VHL requires both the TRiC complex and Hsp70 for correct folding and function and association of VHL with TRiC/Hsp70 can be detected by co-immunoprecipitation (Melville et al., 2003). To test whether HSF1A inhibits TRiC function in vivo, the interaction between TRiC and VHL was assessed in extracts from cells treated with HSF1A or DMSO solvent, after HA-tagged-VHL immunoprecipitation. In control cells HA-VHL was immunoprecipitated with Hsp70 and the TRiC complex. HSF1A treatment reduced the association of Cct3 and Cct8 with VHL approximately 50% and 80% respectively, while association of Hsp70 with HA-VHL was unaffected (Figure 5C, D).
As HSF1A was identified as an activator of human HSF1 expressed in yeast, and HSF1A- Biotin binds both mammalian and yeast TRiC, humanized HSF1 yeast cells were used to ascertain whether yeast TRiC represses human HSF1. Assembly of the functional TRiC chaperonin is dependent on the correct stoichiometry of the individual subunits. Disruption of the stoichiometry of the TRiC subunits, by overexpression of one subunit, reduces yeast cell viability (Lin et al., 1997). To ascertain if reduced yeast TRiC activity promotes human HSF1 activation in yeast, a strain expressing yeast HSF from a URA3 plasmid, and human HSF1 from a LEU2 plasmid, was used as recipient to over-express individual TRiC subunit genes. Activation of human HSF1 is demonstrated by the ability of cells to grow on medium containing 5-FOA, indicative of the ability of cells to lose the URA3-based plasmid carrying the yeast HSF gene. Over-expression of all five individual TRiC subunits tested promoted human HSF1-dependent yeast growth, suggesting that yeast TRiC represses human HSF1 function in yeast (Figure 6A). Neither HSF1A, nor TRiC subunit over-expression activated expression from a yeast HSF-lacZ fusion reporter gene in a wild type yeast strain, indicating that these conditions did not cause global protein misfolding (Figure 6B, C). Moreover, consistent with the low conservation of primary structure between yeast and human HSF1 (Figure S4A), and the constitutive trimerization state of yeast HSF, these results suggest that yeast HSF is not regulated by TRiC. In addition, the high sequence identity between yeast and mammalian TRiC of ~ 47 to 65% (Figure S4B) is consistent with both yeast and mammalian TRiC being able to bind to human HSF1.
Figure 6. TRiC inhibits human HSF1 activation in yeast and mammalian cells.

(A) S. cerevisiae strain DNY248 expressing human HSF1 (hHSF1) or an empty vector (V) were transformed with plasmids expressing the indicated TRiC subunits and grown on SC-His or SC-His supplemented with 5-FOA. (B) S. cerevisiae strain DNY227 harboring an SSA3-β-galactosidase fusion gene was treated with 25 μM HSF1A or DMSO for 6 h or heat shocked at 39 °C for 3 h. Reporter gene activation was assessed by β-galactosidase activity assays. (C) Yeast strain BY4741 expressing a plasmid-borne SSA3 promoter-β-galactosidase fusion gene was transformed with either an empty vector or plasmids expressing the CCT5 or CCT8 genes. (D) Yeast-based assay scheme for impact of TRiC dysfunction on human HSF1 activation. Shown is the HSF1-LexA fusion protein bound to a LexA operator site upstream of the lacZ gene. See also Figure S4C. (E) Yeast strains BJ2168 (WT) and MA6 (CCT6 D89E) were transformed with a plasmid expressing HSF1-LexA and a LexA operator-dependent β-galactosidase reporter gene (left) or an SSA3-β-galactosidase fusion gene (right). Reporter gene activation was assessed by β-galactosidase activity assays. (F) HeLa cells treated for 72 h with siRNA against TCP1 or CCT3, or scrambled siRNA were analyzed for Hsp70, Tcp1, Cct3 and GAPDH levels by immunoblotting and (G) for Hsp70 levels (ng/mL) by ELISA. (H) NIH3T3 cells were transiently transfected with a plasmid expressing mouse TCP1 (CMV-TCP1) or vector control (VEC) and analyzed for Hsp70, Tcp1 and GAPDH protein levels by immunobloting. (I) NIH3T3 cells were treated as in (H) and Hsp70 transcript levels quantitated by qRT-PCR normalized to GAPDH.
While partial loss of function mutations in TRiC subunits have been described, it is difficult to ascertain the specific contribution of these mutations on the activation of human HSF1 in yeast, since both human HSF1 and yeast HSF bind to similar promoter elements. To circumvent this complexity, yeast were transformed with a plasmid encoding a human HSF1 protein that lacks the DNA binding domain but is fused to the DNA binding domain of the prokaryotic LexA repressor (HSF1-LexA) (Figure 6D). HSF1-LexA binds the LexA operator and lack of activity of HSF1 persists in the HSF1-LexA fusion, as the wild-type fusion protein does not promote activation of a LexA operator, β-galactosidase reporter, while the constitutively activate HSF1S303A mutant (Batista-Nascimento et al., 2011) promotes robust activation of the reporter plasmid (Figure S4C). Expression of HSF1-LexA in a strain expressing a CCT6-D89E mutant allele, which partially disrupts Cct6 function (Amit et al., 2010), resulted in activation of the LexA Op-lacZ reporter as compared to the wild type strain. No activation of the yeast HSF- specific SSA3-lacZ reporter was observed in the CCT6-D89E mutant strain (Figure 6E). These results demonstrate that TRiC functions to repress human HSF1, but not yeast HSF, in yeast. Moreover, these results suggest that TRiC represses human HSF1 independently of its DNA binding function.
To ascertain if TRiC represses mammalian HSF1 in mammalian cells, expression of TRiC subunits was reduced by RNAi and Hsp70 protein and mRNA levels were assessed. As previously reported, knock-down of either TCP1 or CCT3 in HeLa cells (Figure 6F) or 3T3 cells (Figure S5) resulted in significantly diminished expression of the RNAi-targeted gene and other TRiC subunits (Brackley and Grantham, 2010). Knock-down of either TCP1 or CCT3 resulted in a ~2-fold increase in Hsp70 expression in unstressed cells (Figure 6F, G). Similarly, over-expression of TCP1 in 3T3 cells resulted in significant elevation of both Hsp70 protein and mRNA levels, as measured by immunoblotting and qRT-PCR, respectively (Figure 6H, I). These results demonstrate that TRiC represses human HSF1 activity in both yeast and mammalian cells.
HSF1A antagonizes direct inhibition of HSF1 by TRiC
To ascertain if repression of HSF1 by TRiC occurs via TRiC-HSF1 interactions, co- immuno-precipitation experiments were conducted by transfecting HEK293T cells with plasmids to express FLAG-tagged HSF1 protein and cells were treated with or without the membrane permeable cross linker DSP, as interactions between HSF1 and Hsp90 are stabilized by the addition of a cross linker (Zou et al., 1998). FLAG-HSF1 was immunoprecipitated and associated proteins analyzed by immunoblotting. Hsp70 co-purified with HSF1, with some enrichment after treatment with DSP, while Hsp90 was highly enriched upon addition of the cross linker (Figure 7A). Co-purification of the TRiC complex, visualized through immunoblotting for the Cct2 and Cct3 subunits, was preferentially observed in the presence of cross linker, suggesting that, like Hsp90, the TRiC-HSF1 interaction is labile. The DSP-stabilized TRiC-HSF1 interaction was independently validated utilizing the dual carboxyl-terminal TAP-GFP-tagged mouse HSF1 allele expressed in HEK293T cells (Figure 7B). No interaction between HSF1 and the abundant protein tubulin was detected.
Figure 7. TRiC directly interacts with HSF1.

(A) HEK293T cells transfected with a plasmid expressing FLAG-HSF1 or an empty vector (Vec), were treated with DSP (+) or left untreated (−). HSF1 was immunoprecipitated from total protein extract using anti-FLAG-affinity resin and analyzed for HSF1, Hsp90, Hsp70, Cct2 and Cct3 by immunobloting. * indicates non-specific band. (B) HEK293T cells transfected with mouse HSF1-GFP-TAP or an empty vector were treated with DSP and HSF1 from total protein extract using anti-GFP-affinity resin. The immunoprecipitate was analyzed for HSF1, Hsp90, Hsp70, Cct3 by immunoblotting. (I, input; PD, pull-down) (C) Purified bovine TRiC, His6-tagged HSF1ΔLZ1-3 or His6-tagged wild-type HSF1 were incubated either alone or with the indicated combinations. Proteins were captured using cobalt-agarose resin and analyzed for HSF1, Cct2 and Cct3 by immunoblotting. Shown are images from the same blot that was separately probed with the indicated antibodies (D) In vitro TRiC – HSF1 interaction experiment carried out as in (C) but purified TRiC was pre-incubated with 200 μM HSF1A-Biotin or Biotin alone prior to addition of HSF1. (E) NIH3T3 and HEK293T cells transfected with a plasmid expressing FLAG-HSF1 were treated with 80 and 100 μM HSF1A, respectively, or DMSO for 6 h, cross-linked with DSP, HSF1 immunoprecipitated and immunoblotted as in (A) for HSF1, Hsp90, Hsp70 and Cct3. (F) Model for the repressive interaction between TRiC and HSF1 and its modulation by HSF1A.
To test whether TRiC and HSF1 directly interact, purified His6-tagged HSF1 was incubated either alone, or with purified bovine TRiC, and HSF1 was affinity captured by cobalt-resin and analyzed by immunoblotting. While very low levels of TRiC (ascertained by immunoblotting for Cct2 and Cct3) bound to the cobalt-resin (Figure 7C, lane 4), TRiC was enriched when co-incubated with HSF1 (Figure 7C, lane 8). As TRiC interacts with the N17 domain of the Huntingtin protein that is known to form coiled-coils (Fiumara et al., 2010; Tam et al., 2006; Tam et al., 2009) the possibility that the TRiC-HSF1 interaction is mediated via the HSF1 coiled-coil trimerization domain was investigated. However, as shown in Figure 7C (lane 7) the HSF1-TRiC interaction was not abrogated by deletion of the HSF1 trimerization domain (HSF1ΔLZ1-3). To ascertain if HSF1A influences the HSF1-TRiC interaction in vitro, TRiC was pre-incubated with HSF1A-Biotin (AB), or Biotin (B) alone, before the addition of HSF1. While Biotin alone had no effect on the interaction, HSF1A-Biotin inhibited TRiC-HSF1 complex formation (Figure 7D). Similarly, treatment of NIH3T3 and HEK293T cells with HSF1A, prior to DSP crosslinking, reduced the interaction between TRiC and HSF1 in vivo though this was more variable in vivo (Figure 7E). Taken together, these data suggest a model that HSF1A directly binds to TRiC, perhaps destabilizing the interaction between TRiC and HSF1 resulting in the amelioration of TRiC-mediated repression of HSF1 (Figure 7F).
Discussion
Cytoplasmic proteotoxic stress causes the generation of misfolded proteins that lead to cellular dysfunction and apoptosis. While HSF1 is a central stress-responsive transcription factor that undergoes a switch from an inactive monomer to a DNA binding active homo-trimer, little is known about the mechanisms that regulate this transition, and downstream activation steps, in response to cytosolic protein misfolding. Previous studies reported that the Hsp90 and Hsp70 chaperones associate with HSF1 in cell extracts and play an inhibitory role in HSF1 regulation (Shi et al., 1998; Zou et al., 1998). However, neither purified Hsp90, nor Hsp70, has been reconstituted into complexes with HSF1 in vitro to distinguish between a direct, versus indirect regulatory role on HSF1. In contrast, here we reconstitute a direct interaction between TRiC and HSF1 that can be targeted pharmacologically to activate the stress-protective response.
Our previous studies identified HSF1A as an activator of HSF1 in a humanized yeast screen that is also active in diverse metazoan cell types (Neef et al., 2010). While HSF1A neither inhibits nor binds Hsp90, HSF1A-Biotin and HSF1A-FITC interact with the TRiC/CCT chaperonin complex in yeast and mammalian cell lysates and as purified subunits or in the fully assembled, active complex. HSF1A binds to the TRiC complex directly and modulates TRiC-dependent chaperone activity in vivo and in vitro. While small molecule inhibitors have been described for Hsp90 that result in the destabilization of Hsp90 client proteins and the concomitant activation of HSF1, no direct small molecule inhibitors of the TRiC/CCT complex have been previously reported (Neef et al., 2011).
The precise mechanism by which HSF1A modulates TRiC activity is not yet understood. HSF1A may associate with client-bound TRiC or with the apo-form of TRiC, but in either case, HSF1A interacts with the highly conserved TRiC subunits from both S. cerevisiae and mammalian cells (Fig. S4B). Given that HSF1A binds directly to all four of individual CCT subunits evaluated, the chaperonin ATPase domain was a candidate binding site for HSF1A (Hartl and Hayer-Hartl, 2002). However, our experiments suggest that HSF1A neither strongly inhibits TRiC ATPase activity nor competes effectively for ATP binding. Furthermore, mutagenesis experiments suggest that a distinct chaperonin domain, involving the hinge 2 region, may constitute the site to which HSF1A binds. Indeed, it has been described that TRiC activity and stability can be regulated by Vaccinia-related kinase 2 (VRK2) through a mechanism that requires an interaction with the carboxyl-terminal region of the TRiC equatorial domain, without affecting ATP hydrolysis (Kim et al., 2013). Further analysis will be required to characterize the HSF1A binding site and to understand how HSF1A binding alters TRiC structure, function and regulatory interactions with HSF1.
As TRiC directly interacts with HSF1, our data support a direct repressor role for TRiC in regulating HSF1 activity. Perhaps the binding of HSF1A to TRiC competes for a TRiC-HSF1 interaction surface, or initiates a conformational change in TRiC that reduces the affinity for the HSF1-TRiC interaction. An alternative explanation for HSF1A-dependent HSF1 activation via TRiC is that HSF1A may inhibit TRiC activity, leading to the accumulation of misfolded TRiC client proteins that, in turn, stimulate the HSF1-mediated heat shock response. However, neither HSF1A exposure, nor genetic inhibition of TRiC, promotes the activation of yeast HSF which, like its metazoan counterparts, is activated in response to conditions that cause protein misfolding (Verghese et al., 2012). This suggests that the negative regulation of HSF1 by TRiC is a metazoan feature of the stress response. Unlike the high degree of conservation between the yeast and human TRiC subunits, yeast and human HSF1 show little overall protein sequence homology outside of their DNA binding domains (Fig. S4A). Furthermore, given that yeast HSF has been reported to be a constitutive homo-trimer, the lack of conservation of the HSF-TRiC regulatory interaction in yeast is not surprising. While data presented herein suggest that HSF1A antagonizes TRiC-dependent HSF1 repression, our data demonstrating the co-purification of TRiC and HSF1 in vivo and in vitro strongly support a model that TRiC acts directly on HSF1. Furthermore, our data suggest that the highly conserved TRiC subunits from both S. cerevisiae and human (Figure S4B) are able to engage in repressive interactions with human HSF1. The TRiC interactome encompasses many functional classes of cytoplasmic proteins including those involved in the function of the cytoskeleton, DNA replication and repair, cell cycle progression, RNA processing and protein trafficking (Dekker et al., 2008; Yam et al., 2008).
Studies in E. coli demonstrated that the chaperonin GroEL directly binds to and represses the activity of σ32, the prokaryotic proteotoxic stress-responsive transcription initiation factor (Guisbert et al., 2004; Rodriguez et al., 2008). Indeed, σ32 regulation by DnaK, DnaJ and GroE/L provides a sophisticated chaperone-mediated regulatory circuit in which the protein quality control pathway directly integrates and communicates with the stress-responsive transcription machinery. Our work suggests that this direct regulatory relationship between a chaperonin complex and stress-activated transcription factor is conserved in mammals and provides an additional level for regulating stress activation of HSF1. Recent work demonstrating specific thiol oxidation in Hsp70 as an activating signal for HSF1 highlights the sophisticated mechanisms built into chaperone-mediated regulation of HSF1 (Miyata et al., 2012; Wang et al., 2012). As HSF1 is activated by a plethora of proteotoxic stress conditions, perhaps distinct stresses integrate HSF1 activation via the modulation of distinct sets of chaperone repressors. As arsenic is an HSF1 activator that also inhibits TRiC (Pan et al., 2010), it will be interesting to evaluate whether chaperonin inhibition is a key pathway for HSF1 activation by arsenic. Indeed, Pan et al observed that TRiC activity is very sensitive to thiol oxidation in vivo and in vitro, which may resemble the thiol-dependent regulation of HSF1 observed through the action of other chaperones such as Hsp70 (Miyata et al., 2012; Wang et al., 2012).
Human HSF1 is regulated by Hsp70 and Hsp90, either directly or indirectly and our data show that the TRiC complex directly binds and represses HSF1. HSF1 may exist in sub-populations regulated by different protein chaperones, perhaps in a tissue or cell type dependent manner or in response to diverse proteotoxic stimuli. This hypothesis is supported by the finding that RNAi mediated knock down of TRiC subunits in C. elegans elicits muscle-specific activation of HSF1 (Guisbert et al., 2013) and suggests that TRiC may be a primary regulator of HSF1 in myocytes. Interestingly, tissue specific regulation of HSF1 by different protein chaperones may have direct physiological disease relevance. Evidence has shown that the expression of protein chaperones such as Hsp70, as well as the activity of HSF1, is strongly repressed in insulin-resistant tissues in Type 2 diabetes (Kurucz et al., 2002). While the mechanisms underlying the repression of HSF1 and Hsp70 are not understood, proteomics analysis of muscle biopsies revealed that Hsp70 levels were dramatically reduced, yet levels of individual TRiC subunits were significantly elevated, in diabetes (Hwang et al., 2010). The levels of specific chaperones in different tissues or in disease states may determine their contribution toward HSF1 regulation and allow for sophisticated integration of diverse stressful stimuli. A greater understanding of the mechanisms regulating HSF1 activity in tissues and disease states could lead to development of pharmacological interventions targeting HSF1 for specific human conditions.
Experimental procedures
Yeast and mammalian cells, transfections, siRNA
Yeast cell growth conditions are detailed in figure legends. Mammalian cell lines used in this study were human HeLa and HEK293T cells, mouse NIH3T3, wild- type and hsf1−/− MEFs (McMillan et al., 1998) stably transfected with either pcDNA3.1(+)/Zeo or pcDNA3.1(+)/Zeo-hHSF1 and rat INS 832/13 cells. Plasmids used in this study were transfected into cells using Lipofectamine LTX (Invitrogen) following the manufactures guidelines. siRNA against TCP1 and CCT3 was purchased from Dharmacon and 2 nmoles of each siRNA were transfected into HeLa or 3T3 cells using Dharmafect 1. Yeast strains, plasmids, and antibodies are listed in Extended Experimental Procedures.
HSF1A-Biotin affinity-capture experiments
Protein extracts were generated from mammalian, yeast and E. coli cultures using biotin-binding buffer (20 mM HEPES, 5 mM MgCl2, 1 mM EDTA, 100 mM KCl, 0.03% NP-40) supplemented with 1% Trition-X100 and protease inhibitors. Approximately 0.5 mg of protein extract was incubated with 100 μM HSF1A-Biotin for 4 h at 4 °C and HSF1A-Biotin associated proteins captured by with NeutrAvidin Agarose Resin (Pierce). After washing in biotin binding buffer proteins were eluted using 50 μL biotin elution buffer (100 mM Tris, 150 mM NaCl, 0.1 mM EDTA, 2 mM D-biotin), resolved on a 4–20% SDS- PAGE, and immunoblotted. For purified TRiC and Hsp70 analyses, 5 nM protein was incubated in biotin-binding buffer + 0.5% Triton X-100 with 100 μM biotin or 100 μM HSF1A-Biotin for 4 h at 4 °C and captured with NeutrAvidin Resin. For NiNTA purified yeast Tcp1, different concentrations of Tcp1 0.5 μM, 1 mM, 2 mM, 3 mM and 4 mM in 25 mM Hepes pH 7.5, 150 mM NaCl were incubated with 0.5 μM Biotin or HSF1A-Biotin for 4 h at 4 °C and captured with NeutrAvidin Resin.
TRiC subunit expression in E. coli
ORFs of yeast TRiC subunits were PCR amplified and cloned into the E. coli expression vector pT7-FLAG-4 (Sigma), transformed into BL21(DE3) cells and protein expression induced via the addition of 1 mM IPTG for 3 h at 37 °C. Total protein extracts were generated by cell lysis in biotin binding buffer supplemented with 1% Trition X-100 and protease inhibitors.
HSF1-TRiC co-immunoprecipitation assay
HEK293T cells tranfected with the indicated plasmid were crosslinked with DSP, lysed in IP buffer, and 0.5 mg of protein was immunoprecipitated with anti-FLAG-M2 affinity gel (Sigma) for FLAG-HSF1 or anti-GFP agarose resin (Santa Cruz) for mHSF1-TAP. Captured proteins were eluted with Laemmli buffer and analyzed by immunoblotting.
In vitro TRiC – HSF1 binding assays
Recombinant human HSF1 was purified as previously described (Ahn and Thiele, 2003). Purified HSF1 and purified TRiC (5 nM) were incubated either alone or together in biotin binding buffer for 1 h at RT and captured using a cobalt- agarose resin for 90 min at 4 °C. After washing, bound proteins were eluted with buffer supplemented with 500 mM imidazole and analyzed by immunoblotting.
Thermal Denaturation Profiling, Actin Folding, ATPase Assays, ATP- Sepharose Purification, VHL co-immunoprecipitation, LexA-HSF1 Assay
See Extended Experimental Procedures
Supplementary Material
Figure S1 (Related to Figure 1). HSF1A protects cells from stress induced apoptosis (A) NIH 3T3 cells were pre-treated with 50 μM HSF1A for 15 h, before the addition of 40 mM AZC, followed by a 15 h incubation. (B) Hsf1 −/− MEFs were stably transfected with either pcDNA3.1-HSF1 or empty vector pcDNA3.1. Cells were pre-treated with 50 μM HSF1A for 15 h or DMSO as negative control before the addition of 0.4 μg/mL tunicamycin (top panel) or 40 mM AZC (bottom panel), followed by a 15 h incubation. Cell viability was assayed using Cell Titer Glo. (C) INS-1 cells were pretreated with 20 μM HSF1A for 15 h, before the addition of 30 mM Glucose followed by a 3 day incubation. Cell viability was assayed using Cell Titer Glo.
Figure S2 (Related to Figure 2). HSF1A directly interacts with the TRiC complex. (A) Representative ThermoFluor curves used to calculate melting temperatures in Figure 2E. Increasing fluorescence (RFU) indicates unfolding of TRiC as a function of increased temperature. (B) First Derivative of Melting Curves in (A). TRiC melting is defined as the maximum value of the derivative curves. Increasing concentrations of HSF1A-Biotin shift the peak of the derivative curve to lower temperatures. (C) A Coomassie blue stained SDS-PAGE showing the purification of recombinantly expressed His6-Tcp1. Shown are molecular weight markers, soluble extract and extract pellet, the flow through from NiNTA affinity chromatography, three wash fractions and the eluate fraction. (D) Structure of the HSF1A-FITC conjugate, showing each respective structural region, that was synthesized and used in fluorescence anisotropy assays.
Figure S3 (Related to Figure 5B). HSF1A induces HSF1 targets but does not affect TRiC clients. Cct3 and α-tubulin protein levels were quantitated by image analysis from a representative immuno blot shown in Figure 5B. Quantification was carried out using Quantity One software (BioRad) and protein intensities for each protein were normalized to the protein levels obtained under control conditions before addition of HSF1A.
Figure S4 (Related to Figure 6) Eukaryotic conservation of HSF1 and TRiC (A) Illustration of the homology between yeast and human HSF1. While the full length proteins share only 26% identity in amino acid sequence, the DNA binding domain and trimerization domain share 31% identity and 50% homology. (B) Crystal structure of the eukaryotic TRiC/CCT complex (Leitner et al., 2012) depicting the high sequence identity and homology between yeast and human TRiC/CCT. (C) The HSF1-LexA protein is functional in yeast. Yeast strain BJ2168 was transformed with a plasmid expressing LexA, a human HSF1-LexA fusion protein or an HSF1-S303A-LexA fusion protein mutant and a LexA operator-β-galactosidase reporter plasmid. Reporter gene activation was assessed by β-galactosidase activity assays.
Figure S5 (Related to Figure 7). TRiC depletion in NIH3T3 cells activates HSF1. NIH3T3 cells were treated with siRNA specific for TCP1, CCT3 or a scrambled siRNA, for 72 h. Total protein was analyzed by immunoblotting for Hsp70, HSF1, Tcp1, Cct3, actin and GAPDH as a loading control.
Acknowledgments
We thank Amnon Horovitz for yeast strains BJ2168 and MA6, Tso-Pang Yao and Chandra Tucker for plasmids, Tim Haystead for the ATP-resin and advice and David Gooden at the Duke Small Molecule Synthesis Facility for chemical synthesis. This work was supported by grants from the National Institutes of Health (NS065890 to D.J.T), (R01GM74074 to J.F.), NRSA Postdoctoral Fellowship GM076954 (to D.W.N.) and EMBO Long-term Fellowship to F.W. A.M.J. is a trainee of the Duke University Pharmacological Sciences Training Program (NIH 2 T32 GM007105).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes & Development. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit M, Weisberg SJ, Nadler-Holly M, McCormack EA, Feldmesser E, Kaganovich D, Willison KR, Horovitz A. Equivalent mutations in the eight subunits of the chaperonin CCT produce dramatically different cellular and gene expression phenotypes. Journal of Molecular Biology. 2010;401:532–543. doi: 10.1016/j.jmb.2010.06.037. [DOI] [PubMed] [Google Scholar]
- Batista-Nascimento L, Neef DW, Liu PC, Rodrigues-Pousada C, Thiele DJ. Deciphering human heat shock transcription factor 1 regulation via post-translational modification in yeast. PloS one. 2011;6:e15976. doi: 10.1371/journal.pone.0015976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiani M, Daniel C, Liu X, Hogarty MD, Marnett LJ. The stress protein BAG3 stabilizes Mcl-1 protein and promotes survival of cancer cells and resistance to antagonist ABT-737. The Journal of Biological Chemistry. 2013;288:6980–90. doi: 10.1074/jbc.M112.414177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackley KI, Grantham J. Subunits of the chaperonin CCT interact with F-actin and influence cell shape and cytoskeletal assembly. Exp Cell Res. 2010;316:543–553. doi: 10.1016/j.yexcr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, Newman HW, Braun S, Madhani HD, Krogan NJ, Weissman JS. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Meth. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Cotto JJ, Kline M, Morimoto RI. Activation of heat shock factor 1 DNA binding precedes stress-induced serine phosphorylation. Evidence for a multistep pathway of regulation. The Journal of Biological Chemistry. 1996;271:3355–3358. doi: 10.1074/jbc.271.7.3355. [DOI] [PubMed] [Google Scholar]
- Dekker C, Stirling PC, McCormack EA, Filmore H, Paul A, Brost RL, Costanzo M, Boone C, Leroux MR, Willison KR. The interaction network of the chaperonin CCT. EMBO J. 2008;27:1827–1839. doi: 10.1038/emboj.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JS, Haystead TA, Litchfield DW. Chemoproteomic characterization of protein kinase inhibitors using immobilized ATP. Methods in Molecular Biology. 2012;795:119–134. doi: 10.1007/978-1-61779-337-0_8. [DOI] [PubMed] [Google Scholar]
- El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, Hardy S, Joly E, Dbaibo G, Rosenberg L, Prentki M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144:4154–4163. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- Fiumara F, Fioriti L, Kandel ER, Hendrickson WA. Essential Role of Coiled Coils for Aggregation and Activity of Q/N-Rich Prions and PolyQ Proteins. Cell. 2010;143:1121–1135. doi: 10.1016/j.cell.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Takaki E, Hayashi T, Kitaura Y, Tanaka Y, Inouye S, Nakai A. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. The Journal of Biological Chemistry. 2005;280:34908–34916. doi: 10.1074/jbc.M506288200. [DOI] [PubMed] [Google Scholar]
- Glover-Cutter KM, Lin S, Blackwell TK. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genetics. 2013;9:e1003701. doi: 10.1371/journal.pgen.1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalves SE, Moses AM, Razak Z, Robert F, Westwood JT. Whole-genome analysis reveals that active heat shock factor binding sites are mostly associated with non-heat shock genes in Drosophila melanogaster. PloS One. 2011;6:e15934. doi: 10.1371/journal.pone.0015934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisbert E, Czyz DM, Richter K, McMullen PD, Morimoto RI. Identification of a tissue-selective heat shock response regulatory network. PLoS Genetics. 2013;9:e1003466. doi: 10.1371/journal.pgen.1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisbert E, Herman C, Lu CZ, Gross CA. A chaperone network controls the heat shock response in E. coli. Genes & Development. 2004;18:2812–2821. doi: 10.1101/gad.1219204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8:e1000410. doi: 10.1371/journal.pbio.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Molecular and Cellular Biology. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hietakangas V, Ahlskog JK, Jakobsson AM, Hellesuo M, Sahlberg NM, Holmberg CI, Mikhailov A, Palvimo JJ, Pirkkala L, Sistonen L. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Molecular and Cellular Biology. 2003;23:2953–2968. doi: 10.1128/MCB.23.8.2953-2968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, Smoke CC, Meyer C, Hojlund K, Yi Z, et al. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes. 2010;59:33–42. doi: 10.2337/db09-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisman N, Schroder GF, Levitt M. The crystal structures of the eukaryotic chaperonin CCT reveal its functional partitioning. Structure. 2013;21:540–549. doi: 10.1016/j.str.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Park DY, Lee D, Kim W, Jeong YH, Lee J, Chung SK, Ha H, Choi BH, Kim KT. Vaccinia-related kinase 2 mediates accumulation of polyglutamine aggregates via negative regulation of the chaperonin TRiC. Molecular and cellular biology. 2014;34:643–652. doi: 10.1128/MCB.00756-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H, Matsumoto S, Yokota S, Yanagi H, Yura T. Transcriptional activation of mouse cytosolic chaperonin CCT subunit genes by heat shock factors HSF1 and HSF2. FEBS Lett. 1999;461:125–129. doi: 10.1016/s0014-5793(99)01437-4. [DOI] [PubMed] [Google Scholar]
- Kurucz I, Morva A, Vaag A, Eriksson KF, Huang X, Groop L, Koranyi L. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes. 2002;51:1102–1109. doi: 10.2337/diabetes.51.4.1102. [DOI] [PubMed] [Google Scholar]
- Leitner A, Joachimiak LA, Bracher A, Monkemeyer L, Walzthoeni T, Chen B, Pechmann S, Holmes S, Cong Y, Ma B, et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure. 2012;20:814–825. doi: 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Cardillo TS, Richard LM, Segel GB, Sherman F. Analysis of mutationally altered forms of the Cct6 subunit of the chaperonin from Saccharomyces cerevisiae. Genetics. 1997;147:1609–1633. doi: 10.1093/genetics/147.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. The Journal of Biological Chemistry. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- Melville MW, McClellan AJ, Meyer AS, Darveau A, Frydman J. The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel-Lindau tumor suppressor complex. Molecular and Cellular Biology. 2003;23:3141–3151. doi: 10.1128/MCB.23.9.3141-3151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y, Rauch JN, Jinwal UK, Thompson AD, Srinivasan S, Dickey CA, Gestwicki JE. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chemistry & Biology. 2012;19:1391–1399. doi: 10.1016/j.chembiol.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8:e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Reissman S, Douglas NR, Huang Z, Yuan DS, Wang X, McCaffery JM, Frydman J, Boeke JD. Trivalent Arsenic Inhibits the Functions of Chaperonin Complex. Genetics. 2010;186:725–734. doi: 10.1534/genetics.110.117655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- Reissmann S, Joachimiak LA, Chen B, Meyer AS, Nguyen A, Frydman J. A gradient of ATP affinities generates an asymmetric power stroke driving the chaperonin TRIC/CCT folding cycle. Cell Rep. 2012;2:866–877. doi: 10.1016/j.celrep.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F, Arsene-Ploetze F, Rist W, Rudiger S, Schneider-Mergener J, Mayer MP, Bukau B. Molecular basis for regulation of the heat shock transcription factor sigma32 by the DnaK and DnaJ chaperones. Molecular Cell. 2008;32:347–358. doi: 10.1016/j.molcel.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Molecular and Cellular Biology. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte TW, An WG, Neckers LM. Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem Biophys Res Commun. 1997;239:655–659. doi: 10.1006/bbrc.1997.7527. [DOI] [PubMed] [Google Scholar]
- Shi Y, Mosser DD, Morimoto RI. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998;12:654–666. doi: 10.1101/gad.12.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovby F, Krassikoff N, Francke U. Assignment of the gene for cystathionine beta-synthase to human chromosome 21 in somatic cell hybrids. Hum Genet. 1984;65:291–294. doi: 10.1007/BF00286520. [DOI] [PubMed] [Google Scholar]
- Spiess C, Meyer AS, Reissmann S, Frydman J. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends in Cell Biology. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nature Structural & Molecular biology. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasiraman V, Ferreyra RG, Frydman J. Folding assays. Assessing the native conformation of proteins. Methods Mol Biol. 2000a;140:169–177. doi: 10.1385/1-59259-061-6:169. [DOI] [PubMed] [Google Scholar]
- Thulasiraman V, Ferreyra RG, Frydman J. Monitoring actin folding. Purification protocols for labeled proteins and binding to DNase I- sepharose beads. Methods Mol Biol. 2000b;140:161–167. doi: 10.1385/1-59259-061-6:161. [DOI] [PubMed] [Google Scholar]
- Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell. 2004;15:1254–1261. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese J, Abrams J, Wang Y, Morano KA. Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol Mol Biol Rev. 2012;76:115–158. doi: 10.1128/MMBR.05018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang J, Dudman NP, Wilcken DE, Lynch JF. Homocysteine catabolism: levels of 3 enzymes in cultured human vascular endothelium and their relevance to vascular disease. Atherosclerosis. 1992;97:97–106. doi: 10.1016/0021-9150(92)90055-l. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gibney PA, West JD, Morano KA. The yeast Hsp70 Ssa1 is a sensor for activation of the heat shock response by thiol-reactive compounds. Mol Biol Cell. 2012;23:3290–3298. doi: 10.1091/mbc.E12-06-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam AY, Xia Y, Lin HT, Burlingame A, Gerstein M, Frydman J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nature Structural & Molecular biology. 2008;15:1255–1262. doi: 10.1038/nsmb.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Cai Y, Adachi MT, Oshiro S, Aso T, Kaufman RJ, Kitajima S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. The Journal of Biological Chemistry. 2001;276:35867–35874. doi: 10.1074/jbc.M100747200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xue R, Zhang Z, Yang X, Shi H. Palmitic and linoleic acids induce ER stress and apoptosis in hepatoma cells. Lipids Health Dis. 2012;11:1. doi: 10.1186/1476-511X-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (Related to Figure 1). HSF1A protects cells from stress induced apoptosis (A) NIH 3T3 cells were pre-treated with 50 μM HSF1A for 15 h, before the addition of 40 mM AZC, followed by a 15 h incubation. (B) Hsf1 −/− MEFs were stably transfected with either pcDNA3.1-HSF1 or empty vector pcDNA3.1. Cells were pre-treated with 50 μM HSF1A for 15 h or DMSO as negative control before the addition of 0.4 μg/mL tunicamycin (top panel) or 40 mM AZC (bottom panel), followed by a 15 h incubation. Cell viability was assayed using Cell Titer Glo. (C) INS-1 cells were pretreated with 20 μM HSF1A for 15 h, before the addition of 30 mM Glucose followed by a 3 day incubation. Cell viability was assayed using Cell Titer Glo.
Figure S2 (Related to Figure 2). HSF1A directly interacts with the TRiC complex. (A) Representative ThermoFluor curves used to calculate melting temperatures in Figure 2E. Increasing fluorescence (RFU) indicates unfolding of TRiC as a function of increased temperature. (B) First Derivative of Melting Curves in (A). TRiC melting is defined as the maximum value of the derivative curves. Increasing concentrations of HSF1A-Biotin shift the peak of the derivative curve to lower temperatures. (C) A Coomassie blue stained SDS-PAGE showing the purification of recombinantly expressed His6-Tcp1. Shown are molecular weight markers, soluble extract and extract pellet, the flow through from NiNTA affinity chromatography, three wash fractions and the eluate fraction. (D) Structure of the HSF1A-FITC conjugate, showing each respective structural region, that was synthesized and used in fluorescence anisotropy assays.
Figure S3 (Related to Figure 5B). HSF1A induces HSF1 targets but does not affect TRiC clients. Cct3 and α-tubulin protein levels were quantitated by image analysis from a representative immuno blot shown in Figure 5B. Quantification was carried out using Quantity One software (BioRad) and protein intensities for each protein were normalized to the protein levels obtained under control conditions before addition of HSF1A.
Figure S4 (Related to Figure 6) Eukaryotic conservation of HSF1 and TRiC (A) Illustration of the homology between yeast and human HSF1. While the full length proteins share only 26% identity in amino acid sequence, the DNA binding domain and trimerization domain share 31% identity and 50% homology. (B) Crystal structure of the eukaryotic TRiC/CCT complex (Leitner et al., 2012) depicting the high sequence identity and homology between yeast and human TRiC/CCT. (C) The HSF1-LexA protein is functional in yeast. Yeast strain BJ2168 was transformed with a plasmid expressing LexA, a human HSF1-LexA fusion protein or an HSF1-S303A-LexA fusion protein mutant and a LexA operator-β-galactosidase reporter plasmid. Reporter gene activation was assessed by β-galactosidase activity assays.
Figure S5 (Related to Figure 7). TRiC depletion in NIH3T3 cells activates HSF1. NIH3T3 cells were treated with siRNA specific for TCP1, CCT3 or a scrambled siRNA, for 72 h. Total protein was analyzed by immunoblotting for Hsp70, HSF1, Tcp1, Cct3, actin and GAPDH as a loading control.