

Abstract
The 40-year-old association of HLA-B27 with ankylosing spondylitis is one of the best examples of disease association with a hereditary marker. Genomewide association and family studies suggest that other important major histocompatibility complex (MHC) influences are operative in ankylosing spondylitis (AS) susceptibility. HLA-B27 positive hepatitis C individuals are immunologically more efficient in combating viral infections such as HIV-1, hepatitis C, and influenza and less efficient in combating against certain bacteria (and perhaps other organisms) capable of surviving intracellularly. A recent representative population survey of the frequency of HLA-B27 in the USA found a lower frequency of HLA-B27 in older US adults, perhaps reflecting this. Other HLA class I and class II alleles have been implicated in AS susceptibility, the most consistent being HLA-B*40/B60 (B*40:01) but also B14, B15, A*0201, DRB1*04:04, and certain DPA1 and DPB1 alleles. Non-HLA MHC alleles have also been implicated, although many such studies have been inconsistent, likely due to power issues related to the low number of HLA-B27-negative AS patients examined. The best evidence is for major histocompatibility complex class I chain-related gene A (MICA) whose recognition by intestinal epithelial T cells expressing different V-delta-1 gamma/delta TCR further implicates the gut in AS pathogenesis. The HLA class I and class II and other non-HLA allelic associations underscore the importance of T cells in AS pathogenesis.
Keywords: Ankylosing spondylitis, Genetics, HLA genes, HLA-B27, Major histocompatibility complex, MIC-A, Spondylitis, Spondyloarthritis
Introduction
HLA class I genes (HLA-A, HLA-B, and HLA-C) and HLA class-II genes (HLA-DRA1, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and HLA-DPB1) encode cell-surface molecules that play an essential role in the immune defense against intracellular infections and in initiating an immune response to invading pathogens, respectively.
Since the original descriptions of the association of HLA-B27 and spondyloarthritis over 40 years ago [1, 2], this has persisted as one of the best examples of a disease association with a hereditary marker. With the advances in the genetic technology of over the past few decades, up to 34 other genes, largely outside of the major histocompatibility complex (MHC), have been described as operative in ankylosing spondylitis (AS) susceptibility [3]. However, their individual contributions are small, and the MHC remains the dominant force in disease susceptibility. Twin and family studies have suggested that the MHC contributes more than 50 % of the total genetic risk for AS [4, 5] and large SNP typing studies suggest even a larger effect (up to 80 % [3]. However, both suggest that MHC influences in addition to HLA-B27 are operative. Linkage disequilibrium (LD) with HLA-B27 has made it difficult to resolve the extent to which other MHC genes play an independent role. This article will review the contribution of MHC to susceptibility to AS and spondyloarthritis (SpA), including the epidemiology and specific role of HLA—to disease causation as it is currently understood. It will not focus on psoriasis or psoriatic arthritis, which merits an independent review in their own right.
The role of HLA-B27
HLA-B*27 is nearly a requirement for AS susceptibility, present in up to 90 % of patients in most ethnic groups. Moreover, homozygosity for HLA-B27 not only enhances disease risk but also the likelihood of familial aggregation and uveitis [4, 6]. How HLA-B27 influences susceptibility to SpA has eluded investigators for over 40 years; however, recent genetic findings have cast light on this. One theory has been that HLA-B27 presents an “arthritogenic peptide.” Despite extensive search for such, none has been found. In fact, recent data demonstrating gene-to-gene interaction between HLA-B27 and endoplasmic reticulum aminopeptidase I (ERAP 1), an important gene identified previously in AS susceptibility and extensively validated [7, 8], suggest that aberrant peptide presentation may be operative in AS susceptibility [3]. Genetic variants associated with reduced function of ERAP1, and loss of expression of ERAP2, are protective for AS [3]. It is possible that these genes operate in AS by an effect on quantity of HLA class I peptide presentation or a qualitative effect on the peptide repertoire presented. Downregulation of ERAP1 and ERAP2 expression has been shown to reduce cell-surface expression of HLA class I molecules.
Alternatively, it has been suggested that misfolding of nascent HLA-B27 in the ER, leading to ER stress, may be involved in the pathogenesis of AS. A rather unique property of HLA-B27 heavy chains is the tendency toward self-adherence, i.e., homodimer formation due to Cys67 residue on a-1 chain (unique to HLA-B27) resulting in recognition by NK cell receptors [9]. This self-adherence may also result in protein misfolding, resulting in pro-inflammatory unfolded protein response (UPR) [10]. It is also possible that by influencing the quantity of peptide available during HLA-B*27 folding, AS-risk ERAP1 and ERAP2 variants slow the rate of this folding, thereby increasing ER stress.
It has also been shown that HLA-B27-positive individuals have altered intracellular killing in certain infections, suggesting that infection or immune response may act as a trigger of SpA [11].
In the last few years, we have begun to learn how profoundly the microbiome shapes the immune response. As a gene that codes for a protein that presents antigen to induce an immune response and that also regulates positive and negative selection of T cells in the thymus, HLA-B27 almost certainly does have an effect on normal human microbial flora. It is possible that additional properties of HLA-B27, such as dimerization, its effect on the unfolded protein response, or its high sequence identity with bacterially derived proteins, all affect bacterial colonization. However, the vast diversity of gut flora and the rather primitive understanding of this diversity make it difficult to quantify how HLA-B27 alters this flora [12].
HLA-B27 itself is known to be highly polymorphic, with over 116 protein subtypes now recognized (HLA-B*27:01-B*27:117-one subtype, B*27:22 was withdrawn due to having originated from a DNA sequencing error) http://www.ebi.ac.uk/cgi-bin/ipd/imgt/hla/get_allele.cgi. Most of these are extremely rare, and few are of enough frequency to have been associated with or described in patients with AS [13]. These include the “parent” or original HLA-B*27:05 as well as B*27:02 (found in whites of European, Middle East, and Northern African origin) (Fig. 1), B*27:04 (originating from East Asia), B:27:07 (from the Middle East and Southern Asia) , and B:27:14 (described in Eskimos and Native Americans) as well as an African subtype occurring in patients with AS but possibly not associated with AS (HLA-B*27:03) and two subtypes not seen in patients with AS (B*27:09 found initially in Sardinians and B*27:06, seen in Southeast Asians) [13].
Fig. 1.
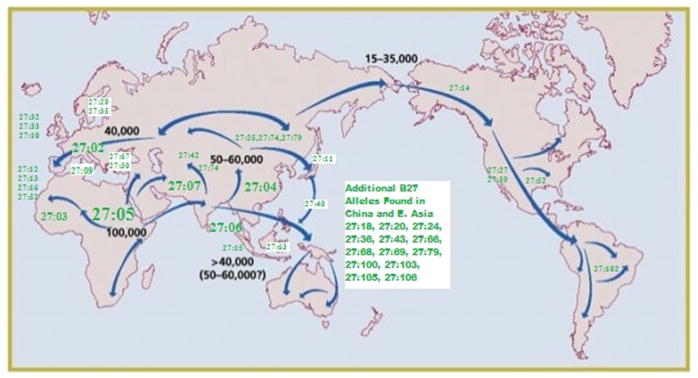
A Map of Prehistoric Human Migrations with Locations of HLA-B27 Subtypes Superimposed. More Common Alleles Are Indication in Larger Fonts
Having HLA-B27 does confer advantages under certain conditions. In the setting of human immunodeficiency virus (HIV) infection, the presence of HLA-B14, B27, B57, and Cw8 slows the progression to AIDS [14]. On the other hand, possessing HLA-A29 and B22 (the latter now split into HLA-B*55 and B*56), as well as two extended HC haplotypes, HLA-A1-Cw7-B8-DR3-DQ2 and HLA-A11, Cw4, B35, DR1, and DQ1 are associated with more rapid progression to AIDS [15, 16] This protective effect of the former alleles has the same magnitude as the CCR5-Delta32 mutation [14]. The mechanisms of this have, at least in part, been elucidated. A study of asymptomatic HIV-infected HLA-B27 and B57-positive individuals found their cytotoxic T lymphocyte response to be dominated by recognition of a gag-encoded p24 protein epitope that was not seen in HLA-B27, B57-negative HIV-positive individuals [17, 18]. In addition, Martin et al. showed that HIV-positive individuals who co-expressed the activating KIR3DS1 allele along with its putative MHC class I ligand Bw4 alleles that contained an isoleucine at position 80 of the peptide-binding groove progressed significantly more slowly toward AIDS than individuals that do not have these two alleles [19]. Although the physical interaction between KIR3DS1 and HLA-Bw4-80I molecules has yet to be shown, this genetic epistasis suggests that this KIR/MHC interaction confers some antiviral signal to NK cells to allow them to control HIV infection more effectively.
HLA-B27 is associated with spontaneous clearance of hepatitis C virus (HCV) infection as well as viral evolution in HCV infection related to a dominant novel CD8+ T cell epitope [20]. HLA-B27 selects for rare escape mutations that significantly impair hepatitis C virus replication and require compensatory mutations [21], and the protective effect is specifically related to the HLA-B*27:02 subtype [22].
There is a downside to having HLA-B27. Individuals positive for this genetic factor have a disadvantage in other infections, particularly in those bacteria able to shed their cell ways and survive intracellularly. Reactive arthritis (ReA) develops as a complication after certain bacterial infections (e.g., Salmonellae, Yersiniae, Shigellae, Campylobacteriae, and Chlamydiae). The risk of ReA in HLA-B27-positive patients has been reported in another study to be up to 50 times higher after the exposure of a triggering infection compared to HLA-B27-negative patients in hospital-based studies. This develops from a complex series of events between the triggering bacteria and the host. A recent review of the distribution of certain infections and HLA alleles shows a negative correlation between the population frequency of HLA-B27 and the prevalence of malaria, another organism capable of surviving intra-cellularly, suggesting that although the high frequency of HLA-B27 in some areas may be explained by a founder effect, there are features to suggest that negative selection by malaria may have contributed to its distribution [23].
In general, the frequency of HLA-B27 in a given population parallels the frequency of SpA. In populations with a high HLA-B27 frequency, the prevalence of AS in particular and SpA is elevated. For example, in the Haida Native Americans, the population frequency of HLA-B27 is 50 %, and the frequency of AS is 6.1 %, whereas in the Japanese, HLA-B27 is rare (less than 1 %, and the prevalence of AS is correspondingly low (0.0065 %) (reviewed in reference 13)). Until recently, there were few actually representative population-based studies of HLA-B27 frequency. The US national prevalence of HLA-B27 was determined as part of the 2009 US National Health and Nutrition Examination Survey (NHANES), a cross-sectional survey monitoring the health and nutritional status of the US civilian, non-institutionalized population [24] (Table 1). The age-adjusted US prevalence of B27 was 6.1 % (Table 1). By race/ethnicity, the prevalence of B27 was 7.5 % among non-Hispanic whites and 3.5 % among all other US races/ethnicities combined. In Mexican Americans, the prevalence was 4.6 %. A lower frequency of HLA-B27 was noted in all groups over the age of 50. Multiple logistic regression analysis of the independent effects of gender, race/ethnicity, and age group showed statistically significantly lower HLA-B27 prevalence estimates for older as opposed to younger US adults (3.6 % for those 50–69 years of age vs. 7.3 % for those 20–49 years, respectively). This begs the question, what is happening to the HLA-B27-positive individuals? Recent as yet unpublished data from China and from the US Veteran’s Administration further confirm these findings, suggesting that HLA-B27-positive individuals in the general population may be at risk for earlier mortality.
Table 1.
The prevalence of HLA-B27 for US adults ages 20–69 years, by selected characteristics, NHANES 2009
| Selected Characteristic | Number | Total number | Percentage | SE | 95 % CI |
|---|---|---|---|---|---|
| Overall US prevalence | 124 | 2,320 | 6.1 | 0.8 | (4.6–8.2) |
| Gender | |||||
| Males | 53 | 1,123 | 5.8 | 1.0 | (3.9–8.4) |
| Females | 71 | 1,197 | 6.5 | 1 | (4.7–8.9) |
| Race/ethnic groups | |||||
| Non-Hispanic Whites | 79 | 1,021 | 7.5 | 1.2 | (5.3–10.4) |
| Mexican-Americans | 27 | 622 | 4.6 | 0.6 | (3.4–6.1) |
| Non-Hispanic Blacks | 4 | 345 | 1.1 | 0.5 | (0.4–3.1) |
| Age groups | |||||
| 20–29 years | 39 | 498 | 8.0 | 2.0 | (4.6–13.4) |
| 30–39 years | 26 | 471 | 5.6 | 1.3 | (3.4–9.2) |
| 40–49 years | 34 | 508 | 8.1 | 1.2 | (5.8–11.2) |
| 50–59 years | 11 | 404 | 2.9 | 0.9 | (1.4–5.8) |
| 60–69 years | 14 | 439 | 4.6 | 1.9 | (1.9–10.7) |
The role of other HLA class I and II alleles
Other HLA-B alleles
Both linkage and association of other MHC class I and II loci with either AS specifically [4] or spondyloarthritis in general [25] have been suggested, but may be confounded due to LD between HLA-B and the MHC class II loci (Table 2). Probably the best characterized and validated association after HLA-B27 is with HLA-B40, most specifically with B:40:01 (a DNA-defined allele that corresponds to HLA-B60 at the serologically defined or protein level) [26]. This association was first described in HLA-B27-positive AS patients from Europe and the USA as far back as 1989 [27], and more recently in the UK and Netherlands [28, 29] HLA-B27-positive patients and in Taiwanese B27-negative patients [30]. HLA-B14 has been associated with AS in French SpA families [25, 31]. Moreover, recent studies from West Africa have shown very low frequencies of HLA-B27 in AS patients; instead, HLA-B*14:03 has been described as being AS-associated [32]. This association has also extended to undifferentiated spondyloarthritis and reactive arthritis in the setting of HIV infection, where association is seen with this as well as with HLA-B*57:03 [33]. The reasons for this association is not entirely clear as the folding, maturation, and stability of B*14:03 differ more from B*27:05 than from disease unassociated B*14:02, suggesting that molecular folding or stability cannot account for this AS association [34]; HLA-B15 has been reported as associated in Mexican patients with peripheral spondyloarthritis [35] as well as in Belgian patients with “undifferentiated SpA” (USpA) [36]. Those with reactive arthritis showed a significant increase with HLA-B15 compared to patients with AS [37]. In Tunisia, HLA-B15 also has been associa
Table 2.
MHC genetic associations with AS other than HLA-B27
| Locus | Function | Associations | Comments | Reference |
|---|---|---|---|---|
| HLA-B (other than B27) | Interact with T-cell receptor molecules as well as killer immunoglobulin-like receptors expressed on natural killer cells and some T cells | All ethnic groups | HLA-B*40:01 (B60) seen in Whites and Asians | [25–38] |
| HLA-A | Interact with T-cell receptor molecules, as well as killer immunoglobulin-like receptors expressed on natural killer cells and some T cells | Whites | Seen in Immunochip Study; small effect | [3] |
| HLA-DRB1 | Encode alpha and beta chains of a heterodimeric, cell-surface glycoprotein that presents antigens to CD4+ (helper) T lymphocytes. | Whites, especially French SpA patients | Needs further study in AS patients. SNP associations in Immunochip Study not suggestive | [25, 38] |
| HLA-DPA1, DPB1 | Encode alpha and beta chains of a heterodimeric, cell-surface glycoprotein that presents antigens to CD4+ (helper) T lymphocytes. | Spanish AS patients | Needs further study in AS patients. SNP associations in Immunochip Study not confirmatory | [40, 41] |
| MICA | Encodes a 383-amino acid polypeptide with a predicted mass of 43 kD and preferential expression in fibroblasts and epithelial cells may have the capacity to bind peptide or other short ligands. | Sardinian, US, and Han Chinese AS patients | Should be studied in other ethnic groups with adequate numbers of HLA-negative AS patients | [43–46] |
| TNF | Multifunctional proinflammatory cytokine secreted predominantly by monocytes/ macrophages that has effects on lipid metabolism, coagulation, insulin resistance, and endothelial function. | German AS patients | Not seen on other cohorts. Association reflects either LD with another nearby gene (MIC-A, etc) | [47] |
| TAP-1 6p21.32 | Translocates peptides from the cytosol to awaiting major histocompatibility complex (MHC) class I molecules in the endoplasmic reticulum | [48, 49] | ||
| LMP2, LMP7 | Amplify specific endopeptidase activities of the proteasome. Favor the production by proteasomes of the types of peptides found on MHC class I molecules, which terminate almost exclusively with hydrophobic or basic residues. | White (Canadian) AS patients | [50] |
HLA-A
Other MHC HLA alleles have also been implicated. Using single-nucleotide polymorphisms (SNPs) to impute HLA alleles in the Immunochip Study, HLA-A*02:01: was found to be associated with AS susceptibility [3] (Fig. 2). The SNP over the HLA-A locus, rs2394250, tags the classical allele HLA-A*02:01 [3]. The association on the HLA-A*02:01 allele was independent of HLA-B*27 genotype, present in both HLA-B27-positive (OR=1.21) and HLA-B27-negative AS patient (OR=1.36). Since no significant correlation was noted between HLA-B*27 and HLA-A*02:01 tag SNPs, this association was not explained linkage disequilibrium with HLA-B27. It is noteworthy that this has not been seen in previous studies, but the relatively low odds ratios associated with this would require larger sample sizes than have been reported to date (Fig. 2).
Fig. 2.
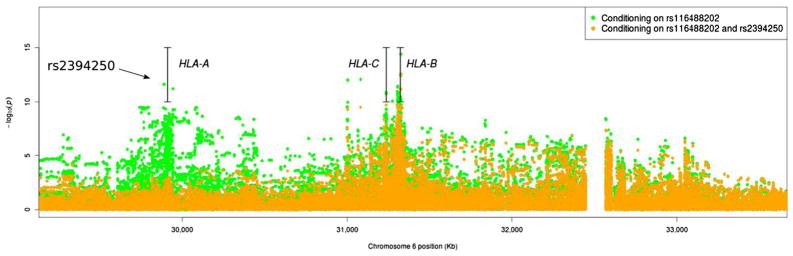
AS susceptibility associations in the MHC locus conditioning on HLA-B*27 tagSNP rs116488202 and further conditioning on HLA-A*02:01 tagSNP rs2394250. The 85-kb gap between positions 32,465 kb and 32,550 kb corresponds to an assembly correction between NCBI human genome build 36 and 37 (the 85-kb gap between positions 32,465 kb and 32,550 kb corresponds to an assembly correction between NCBI human genome build 36 and 37; adapted from reference [3])
DRB1
HLA class II alleles have also been implicated. Early studies from the UK implicated HLA-DRB1*01:01 [4]; however, subsequently, this was attributed to linkage disequilibrium with HLA-B27:05-bearing haplotypes. On the other hand, HLA-DRB1*04:04 was found to be associated in French spondyloarthritis families [25, 38]. While genomewide association studies have not found signals in the MHC class II region [3, 7, 8], it is possible that a small effect of MHC class II alleles may be operative in SpA disease susceptibility.
DQA1
Standard genomewide association studies and direct genotyping of classic HLA alleles do not address the issue of genomic copy number variation (CNV), another source of potential genetic variance. Five AS patients were examined with the high-density comparative genomic hybridization microarrays in the first screen test for AS-associated CNVs. A total of 533 Han Chinese AS patients and 792 unrelated controls examined with the AccuCopy assays after initial resequencing, a significant association, was observed between the CNV of HLA-DQA1 and that of AS compared with controls. AS patients showed an aberrant CN, and a significantly increased number of patients had more than two copies of HLA-DQA1. These are important data, as they suggest that CNV of MHC alleles may have a role in susceptibility to AS [39]. However, no study has shown otherwise any contribution of specific HLA-DWA1 alleles to AS or SpA susceptibility, and these observation will need to be confirmed in other ethnic groups.
DPB1
Located most centromeric of the “classical” MHC alleles, few studies to date have shown the HLA-DPB1 complex. Ploski et al. [40] examined the distribution of B*4001 as well as the DRB1, DPB1, and LMP2 alleles in 63 juvenile onset AS and 44 adult onset AS patients (all B27 positive) finding an increase of the B*40:01, DRB1*08, and DPB1*03:01 alleles as well as the LMP2 b/b genotype (the latter was most pronounced among patients with acute iridocyclitis), in the juvenile onset AS patients compared to B27-positive controls. The increase of DRB1*08 and DPB1*03:01 was due to an increase of DRB1*08 and DPB1*03:01 in combination, whereas the association with B*40:01 could be due to linkage disequilibrium with LMP2b. None of these associations were detected in adult onset AS. Díaz-Peña et al. examined 603 patients with AS and 542 healthy control subjects; all of whom were HLA-B27-positive and found 17 SNPs located within or near the HLA-DPA1 and HLA-DPB1 loci to be associated with AS and confirmed this in a subsequent cohort [41]. Further typing for HLA-DPA1 and HLA-DPB1 alleles found associations with HLA-DPA1*01:03, DPA1*02:01, and DPB1*13:01. However, no association of SNPs around the HLA-DPB1 locus was seen in a much larger cohort of patients examined in the Immunochip Study [3]. Whether this is a phenomenon unique to the Spanish population or represents a true disease association will require further study in other populations.
Other non-HLA MHC genes (Table 2, Fig. 3)
Fig. 3.
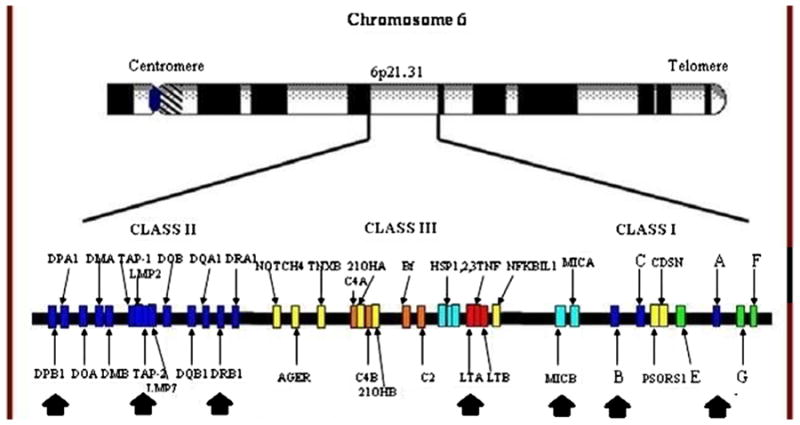
The organization of the MHC, with loci implicated in AS susceptibility indicated by black arrows
MIC-A
The human major histocompatibility complex class I chain-related gene A (MICA) controls the immune process by balancing activities of natural killer cells, γδT cells and αβCD8 Tcells, and immunosuppressive CD4 Tcells. Tcells with variable region V-delta-1 gamma/delta T cell receptor (TCRG, 186,970; TCRD, 186,810) are distributed across the human intestinal epithelium and may function as sentinels that respond to self antigens. MICA, a distant homolog of the MHC class I protein, expression coincides with this location. Groh et al. [42] found that the MICA and the closely related MICBwere recognized by intestinal epithelial T cells expressing different V-delta-1 gamma/delta TCR. These interactions involving domains alpha-1/alpha-2 MICA and MICB but were independent of antigen processing. With intestinal epithelial cell lines, the expression and recognition of MICA and MICB could be stress-induced. Therefore, these molecules in general can regulate the response to protect the V-delta-1 gamma/delta T cells in the epithelium of the intestinal tract.
Goto et al. reported a triplet repeat polymorphism in the transmembrane region within the MICA gene (MICA-A4) closely linked to HLA-B to be associated with AS in 48 B27-positive Caucasian patients [43]. Subsequently, this group examined 162 Japanese AS patients (140 B27-positive and 22 B27-negative patients) and found no differences in the B27-positive and B27-negative patient groups, as compared to the B27-positive and B27-negative control groups, concluding that the higher phenotype frequency of the A4 allele in the patient group was supposed to be due to a strong linkage disequilibrium between the MICA and HLA-B genes [44].
On the other hand, Ricci-Vitiani analyzed the distribution of MICA triplet repeat polymorphism in subjects with the HLA-B*27:09 subtype (not associated with AS) and in subjects with B*27:05 (associated with AS) haplotypes, and compared the distribution of MICA-A alleles in HLA-B27-negative versus HLA-B27+ patients with AS. A high frequency of MICA-A4 allele in HLA-B27-negative patients with AS from Sardinia suggests the presence within this MHC region of a susceptibility factor albeit weaker than B27 [45].
Recent genomewide association studies indicate that genes most strongly linked to ankylosing spondylitis (AS) susceptibility come from the region containing HLA-B and MICA. In fact, the tag SNP used for the conditional analyses in the Immunochip study (rs116488202) was actually located next to MIC-A. Zhou et al. examined 1,543 AS patients and 1,539 controls from two ethnic populations by sequencing MICA and genotyping HLA-B alleles. Initially, 1,070 AS patients and 1,003 controls of European ancestry were used as a discovery cohort, followed by a confirmation cohort of 473 Han Chinese AS patients and 536 controls. Sequencing of MICA identified that MICA*007:01 is a significant risk allele for AS in both Caucasian and Han Chinese populations and that MICA*019 is a major risk allele in Chinese AS patients. Conditional analysis of MICA alleles on HLA-B27 that unshielded LD effect confirmed associations of the MICA alleles with AS. These data suggested that MICA confers strong susceptibility to AS in US white and Han Chinese [46]. It is likely that prior studies may have been hampered by small numbers of HLA-B27-negative patients examined.
TNF
Given the well-described response of AS and SpA to anti-TNF agents, it is entirely reasonable that TNF alleles may play a role is SpA susceptibility. Hohler et al. examined [47] TNF promoter alleles TNF-238.2 and TNF-308.2 in the 141 AS patients, 46 B27-positive and 99 B27-negative controls. Positivity for the variant promoter alleles conferred protection and a relative risk of 0.3 to B27-positive individuals showing data suggesting that variant alleles in the TNFalpha promoter may influence disease susceptibility in HLA-B27-positive individuals. This leads to the irresistible conclusion of a potential protective effect of variant promoter alleles due to differences in TNFalpha production. Alternatively, this could show that different B27 haplotypes are operative in susceptibility to AS [47].
TAP1 and 2
Fraile et al. examined TAP1 and TAP2 alleles in 44 AS subjects and 61 ethnically matched random individuals as well as in 35 B*27-positive healthy unrelated individuals [48]. While the frequency of the TAP1B allele was significantly greater in the patient group compared with the random controls, the results were not significant when HLA-B*27- positive healthy controls were compared, perhaps due to the rather small number of individuals examined. No differences were observed in TAP2 alleles between the groups studied. Likewise, Barron et al. determined TAP1 and TAP2 alleles in 34 patients with reactive arthritis (28 HLA-B27 positive, 6 HLA-B27 negative), and 52 HLA-B27 positive and 80 random disease-free control subjects, finding an association with a specific TAP allele (TAP1C) [49]. These findings have not been confirmed subsequently in other cohorts.
LMP2
Maksymowych et al. examined a polymorphic restriction enzyme site in the coding region of one proteasome gene in B27 individuals with AS, 55 of whom had had iritis and 42 samples from normal, ethnically matched B27 controls without AS. Homozygosity for the disease associated allele was associated with AS with iritis, suggesting an involvement of additional HLA linked genes in the phenotypic expression of disease in B27 individuals and suggest a role for the non-B27 HLA haplotype in susceptibility to iritis [50]. While an interesting observation, it has not been subsequently confirmed in other cohorts.
Summary
Although HLA-B27 remains a dominant risk factor in susceptibility to AS, other important MHC influences are seen in AS susceptibility. Being HLA-B27 positive confers a survival advantage against certain viral infections (HIV, HCV, influenza), perhaps due to interaction with KIR alleles, but a disadvantage against certain bacteria (and perhaps other organisms) capable of surviving intracellularly. Perhaps in part related to this, a representative population survey of HLA-B27 frequency in the USA found a lower frequency of HLA-B27 in older US adults. Other HLA class I and class II alleles have been implicated in AS susceptibility, the most consistent being HLA-B*40/B60 (B*40:01). Non-HLA alleles have also been implicated, although many such studies have been inconsistent, likely due to power issues related to the low number of HLA-B27-negative AS patients examined. The best evidence is for MICA whose recognition by intestinal epithelial T cells expressing different V-delta-1 gamma/delta TCR further implicates the gut in AS pathogenesis. The HLA class I and class II and other non-HLA allelic associations underscore the importance of T cells in AS pathogenesis.
Acknowledgments
This work was supported by the National Institutes of Health-National Institute of Allergy and Infectious Diseases (NIH-NIAIS) grant UO1 AI09090, National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIH-NIAMS) grants R01 AR-46208 and 2P01AR052915-06A1, and by University Clinical Research Grants M01-RR-02558 (The University of Texas Health Science Center at Houston.
Footnotes
Disclosures: None.
References
- 1.Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med. 1973;288:704–706. doi: 10.1056/NEJM197304052881403. [DOI] [PubMed] [Google Scholar]
- 2.Brewerton DA, Hart FD, Nicholls A, et al. Ankylosing spondylitis and HL-A. 27. Lancet. 1973;301(7809):904–907. doi: 10.1016/s0140-6736(73)91360-3. [DOI] [PubMed] [Google Scholar]
- 3.Cortes A, Hadler J, Pointon JP, et al. Multiple novel loci harbouring common and rare variants implicated in ankylosing spondylitis. International Genetics of Ankylosing Spondylitis Consortium (IGAS) Nat Genet. 2013;45:730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown MA, Kennedy LG, MacGregor AJ, et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum. 1997;40:1823–1828. doi: 10.1002/art.1780401015. [DOI] [PubMed] [Google Scholar]
- 5.Zhang G, Luo J, Bruckel J, et al. Genetic studies in familial ankylosing spondylitis susceptibility. Arthritis Rheum. 2004;50:2246–2254. doi: 10.1002/art.20308. [DOI] [PubMed] [Google Scholar]
- 6.Joshi R, Reveille JD, Brown MA, et al. Is there a higher genetic load of susceptibility loci in familial ankylosing spondylitis? Arthritis Care Res (Hoboken) 2012;64:780–784. doi: 10.1002/acr.21601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Australo-Anglo-American Spondyloarthritis Consortium (TASC) and the Wellcome Trust Case Control Consortium 2 (WTCCC2) Genome-wide association study in ankylosing spondylitis identifies further non-MHC associations, and demonstrates that the ERAP1 association is restricted to HLA-B27 positive cases implicating peptide presentation as the likely mechanism underlying the association of HLA-B27 with the disease. Nat Genet. 2011;43:761–767. [Google Scholar]
- 8.WTCCC TASC. Association scan of 14,500 nsSNPs in four common diseases identifies variants involved in autoimmunity. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tam LS, Gu J, Yu D. Pathogenesis of ankylosing spondylitis. Nat Rev Rheumatol. 2010;6:399–405. doi: 10.1038/nrrheum.2010.79. [DOI] [PubMed] [Google Scholar]
- 10.Layh-Schmitt G, Colbert RA. The interleukin-23/interleukin-17 axis in spondyloarthritis. Curr Opin Rheum. 2008;20:392–397. doi: 10.1097/BOR.0b013e328303204b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannu H, Inman RD, Granfors K, Leirisalo-Repo M. Reactive arthritis or postinfectious arthritis. Best Pract Res Clin Rheum. 2006;20:419–433. doi: 10.1016/j.berh.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Rosenbaum JT, Davey MP. Time for a gut check: evidence for the hypothesis that HLA-B27 predisposes to ankylosing spondylitis by altering the microbiome. Arthritis Rheum. 2011;63:3195–3198. doi: 10.1002/art.30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reveille JD, Maganti RM. Subtypes of HLA-B27: history and implications in the pathogenesis of ankylosing spondylitis. Adv Exp Med Biol. 2009;649:159–176. doi: 10.1007/978-1-4419-0298-6_12. [DOI] [PubMed] [Google Scholar]
- 14.Hendel H, Caillat-Zucman S, Lebuanec H, et al. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J Immunol. 1999;162:6942–6946. [PubMed] [Google Scholar]
- 15.Dorak MT, Tang J, Tang S, et al. Influence of human leukocyte antigen-B22 alleles on the course of human immunodeficiency virus type 1 infection in 3 cohorts of white men. J Infect Dis. 2003;188:856–863. doi: 10.1086/378071. [DOI] [PubMed] [Google Scholar]
- 16.Roger M. Influence of host genes on HIV-1 disease progression. FASEB J. 1998;12:625–632. doi: 10.1096/fasebj.12.9.625. [DOI] [PubMed] [Google Scholar]
- 17.Goulder PJ, Phillips RE, Colbert RA, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 18.Streeck H, Lichterfeld M, Alter G, et al. Recognition of a defined region within p24 gag by CD8+ T cells during primary human immunodeficiency virus type 1 infection in individuals expressing protective HLA class I alleles. J Virol. 2007;81:7725–7731. doi: 10.1128/JVI.00708-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin MP, Gao X, Lee JH, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 20.Neumann-Haefelin C. HLA-B27-mediated protection in HIV and hepatitis C virus infection and pathogenesis in spondyloarthritis: two sides of the same coin? Curr Opin Rheumatol. 2013;25:426–433. doi: 10.1097/BOR.0b013e328362018f. [DOI] [PubMed] [Google Scholar]
- 21.Neumann-Haefelin C, Oniangue-Ndza C, Kuntzen T, et al. Human leukocyte antigen B27 selects for rare escape mutations that significantly impair hepatitis C virus replication and require compensatory mutations. Hepatology. 2011;54:1157–1166. doi: 10.1002/hep.24541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nitschke K, Barriga A, Schmidt J, et al. HLA-B*27 subtype specificity determines targeting and viral evolution of a hepatitis C virus-specific CD8+ T cell epitope. J Hepatol. 2014;60:22–29. doi: 10.1016/j.jhep.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathieu A, Paladini F, Vacca A, et al. The interplay between the geographic distribution of HLA-B27 alleles and their role in infectious and autoimmune diseases: a unifying hypothesis. Autoimmun Rev. 2009;8(5):420–425. doi: 10.1016/j.autrev.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Reveille JD, Hirsch R, Dillon CF, Carroll MD, Weisman MH. The Prevalence of HLA-B27 in the United States: Data from the U.S. National Health and Nutrition Examination Survey, 2009. Arthritis Rheum. 2012;64:1407–1411. doi: 10.1002/art.33503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breban M. Genetic studies of spondylarthropathies. French Spondylarthropathy Genetic Study Group. Ann Med Interne (Paris) 1998;149:142–144. [PubMed] [Google Scholar]
- 26.Kawaguchi G, Kato N, Kashiwase K, et al. Structural analysis of HLA-B40 epitopes. Hum Immunol. 1993;36:193–198. doi: 10.1016/0198-8859(93)90125-k. [DOI] [PubMed] [Google Scholar]
- 27.Robinson WP, van der Linden SM, Khan MA, et al. HLA-Bw60 increases susceptibility to ankylosing spondylitis in HLA-B27+ patients. Arthritis Rheum. 1989;32:1135–1141. doi: 10.1002/anr.1780320912. [DOI] [PubMed] [Google Scholar]
- 28.Brown MA, Pile KD, Kennedy LG, et al. HLA class I associations of ankylosing spondylitis in the white population in the United Kingdom. Ann Rheum Dis. 1996;55:268–270. doi: 10.1136/ard.55.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Gaalen FA, Verduijn W, Roelen DL, et al. Epistasis between two HLA antigens defines a subset of individuals at a very high risk for ankylosing spondylitis. Ann Rheum Dis. 2013;72:974–978. doi: 10.1136/annrheumdis-2012-201774. [DOI] [PubMed] [Google Scholar]
- 30.Wei JC, Tsai WC, Lin HS, Tsai CY, Chou CT. HLA-B60 and B61 are strongly associated with ankylosing spondylitis in HLA-B27-negative Taiwan Chinese patients. Rheumatology (Oxford) 2004;43:839–842. doi: 10.1093/rheumatology/keh193. [DOI] [PubMed] [Google Scholar]
- 31.López-Larrea C, Mijiyawa M, González S, et al. Association of ankylosing spondylitis with HLA-B*1403 in a West African population. Arthritis Rheum. 2002;46:2968–2971. doi: 10.1002/art.10584. [DOI] [PubMed] [Google Scholar]
- 32.Díaz-Peña R, Blanco-Gelaz MA, Njobvu P, et al. Influence of HLA-B*5703 and HLA-B*1403 on susceptibility to spondyloarthropathies in the Zambian population. J Rheumatol. 2008;35:2236–2240. doi: 10.3899/jrheum.080395. [DOI] [PubMed] [Google Scholar]
- 33.Merino Galocha B, Vázquez MN, López de Castro JA. Disparate folding and stability of the ankylosing spondylitis-associated HLA-B*1403 and B*2705 proteins. Arthritis Rheum. 2008;58:3693–3704. doi: 10.1002/art.24045. [DOI] [PubMed] [Google Scholar]
- 34.Vargas-Alarcón G, Hernández-Pacheco G, Pacheco-Tena C, et al. Effect of HLA-B and HLA-DR genes on susceptibility to and severity of spondyloarthropathies in Mexican patients. Ann Rheum Dis. 2002;61:714–717. doi: 10.1136/ard.61.8.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mielants, Veys EM, Joos R, Noens L, Cuvelier C, De Vos M. HLA antigens in seronegative spondyloarthropathies. Reactive arthritis and arthritis in ankylosing spondylitis. Relation to gut Inflammation. J Rheumatol. 1987;14:466–471. [PubMed] [Google Scholar]
- 36.Mielants, Veys EM, De vos M, Cuvelier C, et al. The evolution of spondyloarthropathies in relation to gut histology. I. Clinical aspects. J Rheumatol. 1995;22:2266–2272. [PubMed] [Google Scholar]
- 37.Siala M, Mahfoudh N, Fourati H, et al. MHC class I and class II genes in Tunisian patients with reactive and undifferentiated arthritis. Clin Exp Rheumatol. 2009;27:208–213. [PubMed] [Google Scholar]
- 38.Said-Nahal R, Miceli-Richard C, Gautreau C, et al. The role of HLA genes in familial spondyloarthropathy: a comprehensive study of 70 multiplex families. Ann Rheum Dis. 2002;61:201–206. doi: 10.1136/ard.61.3.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Yang Y, Guo S, et al. Association between copy number variations of HLA-DQA1 and ankylosing spondylitis in the Chinese Han population. Genes Immun. 2013;14:500–503. doi: 10.1038/gene.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ploski R, Flato B, Vinje O, et al. Association to HLA-DRB1*08, HLA-DPB1*0301 and homozygosity for an HLA-linked proteasome gene in juvenile ankylosing spondylitis. Hum Immunol. 1995;44:88–96. doi: 10.1016/0198-8859(95)00063-a. [DOI] [PubMed] [Google Scholar]
- 41.Díaz-Peña R, Aransay AM, Bruges-Armas J, et al. Fine mapping of a major histocompatibility complex in ankylosing spondylitis: association of the HLA-DPA1 and HLA-DPB1 regions. Arthritis Rheum. 2011;63:3305–3312. doi: 10.1002/art.30555. [DOI] [PubMed] [Google Scholar]
- 42.Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial gamma-delta T cells. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- 43.Goto K, Ota M, Ohno S, et al. MICA gene and anky losing spondylitis: linkage analysis via a transmembrane-encoded triplet repeat polymorphism. Tissue Antigens. 1997;49:503–507. doi: 10.1111/j.1399-0039.1997.tb02786.x. [DOI] [PubMed] [Google Scholar]
- 44.Yabuki K, Ota M, Goto K, et al. Triplet repeat polymorphism in the MICA gene in HLA-B27 positive and negative Caucasian patients with ankylosing spondylitis. Hum Immunol. 1999;60:83–86. doi: 10.1016/s0198-8859(98)00092-5. [DOI] [PubMed] [Google Scholar]
- 45.Ricci-Vitiani L, Vacca A, Potolicchio I, et al. MICA gene triplet repeat polymorphism in patients with HLA-B27 positive and negative ankylosing spondylitis from Sardinia. J Rheumatol. 2000;27:2193–2197. [PubMed] [Google Scholar]
- 46.Zhou X, Wang J, Zou H, et al. MICA, a gene contributing strong susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2013 Jun 1; doi: 10.1136/annrheumdis-2013-203352. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hohler T, Schaper T, Schneider PM, et al. Association of different tumor necrosis factor alpha promoter allele frequencies with ankylosing spondylitis in HLA-B27 positive individuals. Arthritis Rheum. 1998;41:1489–1492. doi: 10.1002/1529-0131(199808)41:8<1489::AID-ART20>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 48.Fraile A, Collado MD, Mataran L, et al. TAP1 and TAP2 polymorphism in Spanish patients with ankylosing spondylitis. Exp Clin Immunogenet. 2000;17:199–204. doi: 10.1159/000019139. [DOI] [PubMed] [Google Scholar]
- 49.Barron KS, Reveille JD, Carrington M, Mann DL, Robinson MA. Susceptibility to Reiter's syndrome is associated with alleles of TAP genes. Arthritis Rheum. 1995;38:684–689. doi: 10.1002/art.1780380517. [DOI] [PubMed] [Google Scholar]
- 50.Maksymowych WP, Wessler A, Schmitt-Egenolf M, et al. Polymorphism in an HLA linked proteasome gene influences phenotypic expression of disease in HLA-B27 positive individuals. J Rheumatol. 1997;21:665–669. [PubMed] [Google Scholar]