

Abstract
Trace metal homeostasis is tightly controlled in the brain, as even a slight dysregulation may severely impact normal brain function. This is especially apparent in Alzheimer’s disease, where brain homeostasis of trace metals such as copper and iron is dysregulated. As it is known that iron and copper metabolism are linked, we wanted to investigate if a common mechanism could explain the increase in iron and decrease in copper seen in Alzheimer’s disease brain. Amyloid precursor protein has been implicated in copper efflux from the brain. Furthermore, it was shown that iron regulatory proteins, which regulate iron homeostasis, can block amyloid precursor protein mRNA translation. In a correlative study we have therefore compared brain regional copper levels and AβPP expression in mice with a targeted deletion of iron regulatory protein 2 (IRP2−/−). Compared with controls, six week old IRP2−/− mice had significantly less brain copper in the parietal cortex, hippocampus, ventral striatum, thalamus, hypothalamus and whole brain, while amyloid precursor protein was significantly upregulated in the hippocampus (p<0.05) and showed a trend toward upregulation in the thalamus (p<0.1). This is the first study to demonstrate that iron regulatory proteins affect brain copper levels, which has significant implications for neurodegenerative diseases.
Keywords: IRP2, AβPP, APP, amyloid precursor protein, Alzheimer’s disease
INTRODUCTION
Both copper and iron are trace metals essential for the catalytic activity of a multitude of enzymes involved in critical biologic processes such as antioxidant defense, mitochondrial respiration, and the synthesis of melanin, catecholamines and several neurotransmitters. The tissue concentrations of both metals are tightly regulated as either in the free form can catalyze the generation of reactive oxygen species (ROS). In fact, one study has shown that there is essentially no “free” copper inside the cell [1].
Copper chaperones play an important part in this control of intracellular copper distribution by delivering copper to specific proteins, intracellular compartments and, in case of copper overload, facilitating copper efflux. Two critical copper chaperones are copper chaperone for SOD1 (CCS), which inserts a copper moiety into SOD1 activating an important antioxidant pathway [1], and antioxidant protein 1 (Atx1), which is thought to deliver copper to the Menkes and Wilson disease proteins [2]. Additionally, one of the key proteins of Alzheimer’s disease (AD), amyloid precursor protein (AβPP), has been found to have two binding domains for copper – copper binding domain 1 (CuBD-1) in the N-terminal region and copper binding domain 2 (CuBD-2) in the C-terminal within the Aβ sequence [3–5]. Moreover, CuBD-1 has structural homology to copper chaperones such as CCS and Atx1 [6]. Functionally, AβPP has been implicated in the efflux of copper from cells and the brain [7]. This was demonstrated in several studies using transgenic mice that overexpress human AβPP, which resulted in brain copper to be decreased, as well as AβPP knockout mice, where higher levels of brain copper were found [8–12].
Iron regulatory proteins (IRPs) control the cellular uptake and storage of iron by facilitating or inhibiting the translation of proteins involved in iron metabolism. Of the two IRPs that have been identified, IRP1 and IRP2, IRP2 is the dominant regulator of iron homeostasis in most tissues under physiologic conditions [13,14]. In the absence of iron, IRPs bind to iron response elements (IREs) in the mRNA of iron metabolism proteins. If the IRE lies in the 5′-untranslated region (UTR), IRP binding blocks translation, while IRP binding to an IRE in the 3′-UTR has the opposite effect, stabilizing the mRNA and thereby increasing translation. In the presence of high intracellular iron, IRP2 is degraded and IRP1 functions as a cytosolic aconitase, which abolishes binding to the IREs. In 2002, Rogers et al. reported the discovery of a functional IRE within the 5′-UTR of the AβPP mRNA which shares similarities with the IRE found in the 5′-UTR of ferritin mRNA [15]. Furthermore, they were able to show that AβPP expression is regulated by the presence or absence of iron, an observation later replicated by Reznichenko et al. [16].
The linkage between copper and iron metabolism was detailed in 1928 by Hart et al [17]. but was, in fact, first discovered in the mid-19th century by Millon [18] (For a comprehensive review on the history and relationship between iron and copper homeostasis see Fox [19]). The relationship of iron and copper has been studied extensively, albeit with focus on the influence of copper on iron homeostasis. However, some studies have indicated a role of iron in copper homeostasis, including the effects of iron overload or anemia on serum ceruloplasmin levels [20,21] or the increased expression of the Menkes copper ATPase (ATP7A) in the duodenum and jejunum of iron deficient rats [22].
Here, we propose a mechanism by which increased iron mediates the efflux of copper from the brain, through the effect of IRP2 on AβPP (Figure 1). High levels of iron cause the degradation of IRP2, freeing the IRE in the 5′-UTR of AβPP mRNA for translation. This leads to increased expression of AβPP, which in turn causes a decrease in brain copper. To study this mechanism we have measured total copper and AβPP levels in 12 brain regions in 6, 12 and 24 weeks old control C57BL/6 mice and mice with a targeted deletion of IRP2 (IRP2−/−), which show a tissue specific dysregulation of iron metabolism and are reported to develop neurodegenerative symptoms that worsen with age [23].
Figure 1.
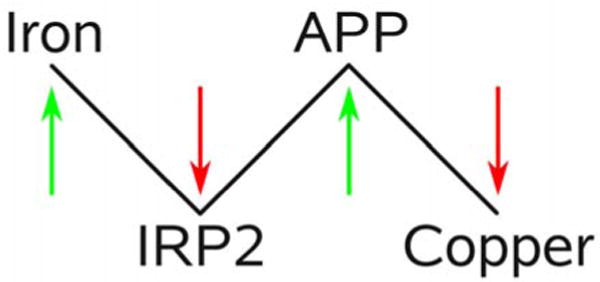
Proposed mechanism of iron mediated copper efflux from the brain. Increased levels of iron lead to the degradation of IRP2. This frees the IRE at the 5′-UTR of the AβPP mRNA and thereby increases expression of AβPP. Higher levels of AβPP in turn then lead to increased efflux of copper from the brain.
MATERIALS AND METHODS
Animals
Male IRP2 knockout mice (IRP2−/−) were bred at Taconic Laboratories (Germantown, NY) from the strain generated and provided by TA Rouault [23]. As their genetic background is a mix of C57BL/6J and B129S4/SVJae in undefined proportions we chose male C57BL/6J (Jackson Laboratories, Bar Harbor, ME) as genetic controls and male C57BL/6NTac (bred at Taconic Laboratories) to control for husbandry. Mice were shipped to our local animal care facility at 5.5±0.5 (SD), 8.9±0.3 and 11±2.7 weeks of age and maintained until they reached the designated ages of either 6, 12 or 24 weeks. Mice were housed according to standard animal care procedures with water and chow provided ad libitum. The copper content of chow at Taconic Laboratories, Jackson Laboratories and our local animal care facility is 17, 12 and 12 mg/kg respectively. Experimental protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Preparation of brain tissue
Mouse brain preparation and regional dissection was performed as previously described [24]. Briefly, mice were anesthetized with Nembutal and brains fixed with focused microwave irradiation for 1 s at 5 kW (Muromachi Microwave Fixation System, Model TMW-6402C, Tokyo, Japan) to inactivate tissue enzymes. Brains were dissected into the following 12 regions: olfactory bulb (OB), frontal cortex (FC), parietal cortex (PC), cerebellum (CB), hippocampus (HP), dorsal striatum (DS), ventral striatum (VS), septum (SP), thalamus (TM), hypothalamus (HY), entorhinal cortex (EC) and brainstem (BS). Immediately following, tissues were weighed (wet weight) and processed for total copper extraction or frozen at −80 °C for later protein extraction. Brain regions were identified according to the mouse brain atlas of Paxinos and Franklin [25].
Copper analysis
Total tissue copper was extracted using the wet ashing method of Maynard et al. with slight modifications [9]. Brain tissue was first dissolved in 300 μl of 70 % nitric acid overnight and then heated at 80 °C for 20 minutes. After cooling to room temperature samples were incubated with 300 μl of 10 M hydrogen peroxide for 30 minutes to dissolve lipid components, after which samples were heated again at 70 °C for 15 minutes and stored at −20 °C until measurement. Copper concentrations were measured at least in duplicate by graphite furnace atomic absorption spectrometry with a SpectrAA 2202Z (Varian, Victoria, Australia). Whole brain copper concentration was determined by summing the amount of copper found in every brain region and dividing it by the sum of brain region weights. A total of 6 to 9 animals were analyzed per brain region, mouse strain and age (whole brain: n = 4–8).
Amyloid precursor protein analysis
Expression of AβPP was analyzed semi-quantitatively using Western blotting. Total protein was extracted by homogenizing brain regions for 20 seconds with polypropylene mortars and pestles (Kontes, Vineland, NJ) in two times the tissue weight of ice cold homogenization buffer (adapted from Levites et al. [26]: 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 300 mM sucrose, 2 mM EDTA, 1 complete Mini protease inhibitor tablet (Roche) per 10 ml of buffer). Crude nuclear material and cell debris were removed by centrifugation at 5000 g at 4 °C for 20 minutes. Supernatants were then frozen at −80 °C until further use. To measure whole brain AβPP expression protein extracts from all regions were mixed according to sample volume and brain region weight. Protein concentration was estimated using the Coomassie Plus Protein Assay (Pierce). Five micrograms of protein were loaded onto a 4–12 % Bis-Tris SDS-PAGE gel (Invitrogen) and transferred to a nitrocellulose membrane (Schleicher & Schuell BioScience) after electrophoresis. AβPP and β-actin were detected using the monoclonal antibodies 22C11 (Chemicon) and AC-15 (Sigma) respectively. The antibody used for the detection of AβPP binds to the N-terminus of the protein and recognizes full length and soluble forms of AβPP [27]. Goat anti-Mouse IRDye800CW (Li-cor) was used as secondary antibody. Protein bands were visualized using the Odyssey Infrared Imaging System (Li-cor). A total of 5 to 7 animals were analyzed per brain region, mouse strain and age (whole brain: 2 to 4 animals per mouse strain and age).
Statistical analysis
Copper concentrations were analyzed using one-way ANOVA and corrected for multiple comparisons using the Bonferroni post-hoc test (SPSS 16). Prior to the ANOVA normality and homogeneity of variances of the data were evaluated. In case of a failed test for normality (Kolmogorov-Smirnov) or of homogeneity of variances data were logarithmic transformed. Upon further failing of either test results were analyzed using the non-parametric Mann-Whitney test.
AβPP expression data were analyzed using the Student′s t-test. Levene′s Test of Equality of Variances and the Kolmogorov-Smirnov test were used to evaluate variance and normality of the data, respectively. If results failed to pass either test even after logarithmic transformation, data were analyzed using the Mann-Whitney non-parametric test. A p value < 0.05 was chosen to indicate significance.
RESULTS
Copper levels in IRP2−/− and control mice
We have measured copper levels in 12 brain regions of mice with a targeted deletion of IRP2 (IRP2−/−) and two control mouse substrains (C57BL6/NTac and C57BL6/J) at 6, 12 and 24 weeks of age. In both control mouse substrains copper levels increased significantly with development in several brain regions, including the olfactory bulb, cerebellum, dorsal striatum, septum, hypothalamus and brainstem (Table 1). This observation was further confirmed in whole brain where significantly higher copper levels were found at 24 weeks compared to 6 or 12 weeks.
Table 1.
Brain copper levels.
| Brain region | Age | IRP2−/− | BL6/NTac | BL6/J | Brain region | Age | IRP2−/− | BL6/NTac | BL6/J |
|---|---|---|---|---|---|---|---|---|---|
| [weeks] | [ug Cu/g wet weight] | [weeks] | [ug Cu/g wet weight] | ||||||
| OB | 6 | 4.1 ± 0.3 | 4.1 ± 0.2 | 4.0 ± 0.4 | SP | 6 | 4.7 ± 0.7 | 5.1 ± 0.7 | 4.5 ± 0.3 |
| 12 | 4.7 ± 0.3 | 5.5 ± 0.4a | 5.5 ± 0.3a | 12 | 6.9 ± 0.5 | 6.4 ± 0.4 | 9.2 ± 2.1 | ||
| 24 | 5.7 ± 0.4b | 6.1 ± 0.6b | 7.1 ± 0.4b,c | 24 | 8.7 ± 1.0b | 11.1 ± 0.7b,c | 13.0 ± 1.4b | ||
| FC | 6 | 3.6 ± 0.1 | 4.1 ± 0.2 | 3.9 ± 0.1 | TH | 6 | 3.8 ± 0.2*,† | 5.0 ± 0.3 | 4.6 ± 0.3 |
| 12 | 4.7 ± 0.6 | 4.4 ± 0.2 | 4.8 ± 0.2a | 12 | 4.9 ± 0.3a | 4.8 ± 0.2 | 5.0 ± 0.2 | ||
| 24 | 5.2 ± 0.2#,b | 4.8 ± 0.3$ | 6.5 ± 0.3b,c | 24 | 7.5 ± 0.9b,c | 6.9 ± 0.9 | 9.0 ± 1.5 b,c | ||
| PC | 6 | 3.8 ± 0.1*,#,† | 4.5 ± 0.2 | 4.3 ± 0.1 | HY | 6 | 3.2 ± 0.1*,#,† | 4.0 ± 0.3 | 5.2 ± 0.8 |
| 12 | 6.0 ± 0.9a | 4.6 ± 0.2 | 5.6 ± 0.4 | 12 | 5.0 ± 1.3 | 4.0 ± 0.2$ | 4.6 ± 0.2 | ||
| 24 | 7.0 ± 1.2b | 5.2 ± 0.3 | 8.6 ± 1.9b,c | 24 | 6.8 ± 1.0b | 5.6 ± 0.9 | 6.9 ± 1.0 | ||
| CB | 6 | 5.1 ± 1.0 | 5.7 ± 0.6 | 5.9 ± 0.5 | EC | 6 | 2.8 ± 0.1 | 3.2 ± 0.3 | 3.0 ± 0.1 |
| 12 | 5.5 ± 0.2# | 5.7 ± 0.2$ | 6.5 ± 0.1 | 12 | 4.4 ± 0.6a | 3.8 ± 0.3 | 5.1 ± 0.5a | ||
| 24 | 7.1 ± 0.3# | 7.9 ± 0.5b,c | 9.2 ± 0.4b,c | 24 | 5.2 ± 1.0b | 3.8 ± 0.1$ | 5.0 ± 0.3b | ||
| HP | 6 | 3.4 ± 0.1*,† | 4.9 ± 0.4 | 4.2 ± 0.2 | BS | 6 | 4.1 ± 0.7 | 4.5 ± 0.4 | 3.8 ± 0.2 |
| 12 | 4.6 ± 0.2a | 5.1 ± 0.8 | 4.9 ± 0.1a | 12 | 4.1 ± 0.2 | 4.1 ± 0.1 | 4.7 ± 0.2a | ||
| 24 | 7.3 ± 1.3b,c | 8.6 ± 3.6 | 8.9 ± 2.2b,c | 24 | 5.0 ± 0.6 | 5.5 ± 0.4 | 6.3 ± 0.5 b,c | ||
| DS | 6 | 4.2 ± 0.3 | 9.3 ± 4.7 | 4.8 ± 0.2 | |||||
| 12 | 6.3 ± 0.8 | 5.9 ± 0.5 | 6.3 ± 0.2 | ||||||
| 24 | 7.6 ± 1.6b | 7.5 ± 0.6$ | 14.5 ± 5.9b,c | ||||||
| VS | 6 | 3.5 ± 0.4 | 4.1 ± 0.3 | 4.0 ± 0.1 | Whole brain | 6 | 3.8 ± 0.2*,#,† | 4.6 ± 0.3 | 4.4 ± 0.1 |
| 12 | 5.0 ± 1.1 | 4.1 ± 0.3 | 4.8 ± 0.2 | 12 | 5.0 ± 0.4a | 4.7 ± 0.1$ | 5.4 ± 0.1a | ||
| 24 | 5.1 ± 0.7# | 5.0 ± 0.3$ | 8.3 ± 1.0b,c | 24 | 6.3 ± 0.6b,c | 6.2 ± 0.4$,b,c | 8.1 ± 0.8b,c | ||
OB = olfactory bulb, FC = frontal cortex, PC = parietal cortex, CB = cerebellum, HP = hippocampus, DS = dorsal striatum, VS = ventral striatum, SP = septum, TH = thalamus, HY = hypothalamus, EC = entorhinal cortex, BS = brainstem (n = 6–9 per brain region, mouse strain and age, whole brain: n = 4–8). Errors represent SEM;
p<0.05 IRP2−/− versus control (C57BL6/J and C57BL6/NTac combined)
p<0.05 IRP2−/− versus C57BL6/J
p<0.05 IRP2−/− versus C57BL6/NTac
p<0.05 C57BL6/NTac versus C57BL6/J
p<0.05 6 versus 12 weeks
p<0.05 6 versus 24 weeks
p<0.05 12 versus 24 weeks.
Some differences in copper levels were observed between the control substrains, with C57BL/6J mice consistently showing higher levels. At 12 weeks copper was elevated in cerebellum, ventral striatum, hypothalamus and whole brain, while at 24 weeks copper was increased in frontal cortex, parietal cortex, ventral striatum and entorhinal cortex compared to C57BL/6NTac mice (Table 1). As no difference was seen between substrains at 6 weeks, the observed copper increase in C57BL/6J compared to C57BL/6NTac mice appears to arise from a more rapid copper accumulation rate in C57BL/6J mice with development. This is further supported by the fact that a significant increase of brain copper with development in frontal cortex, parietal cortex, hippocampus, thalamus and entorhinal cortex was found in C57BL/6J but not C57BL/6NTac mice.
In both C57BL/6 mouse substrains the lowest concentration of copper at 6 weeks was found in the entorhinal cortex, whereas the cerebellum contained the highest concentration of copper, which was about twice as much. By 24 weeks the entorhinal cortex was still the brain region with the least amount of copper present. However, the septum, which had the highest rate of copper increase with development (by a factor of 2.7 from 6 weeks to 24 weeks for both control mouse substrains combined) also had the highest copper concentration at 24 weeks. The smallest increase in copper between 6 and 24 weeks was seen in the hypothalamus (by a factor of 1.2 for both control mouse substrains combined).
A general increase in brain regional copper levels was also observed in IRP2−/− mice. Every IRP2−/− mouse brain region as well as whole brain showed a significant increase in copper levels with development, except for the dorsal striatum (Table 1). Similar to C57BL/6 controls, at 6 weeks copper concentrations were lowest in the entorhinal cortex and highest in the cerebellum in IRP2−/− mice. However, at 24 weeks the lowest concentration of copper in IRP2−/− mice was found in the ventral striatum, as opposed to the entorhinal cortex in control mouse substrains. The highest concentration at that age was again found in the septum.
While the smallest increase in copper in IRP2−/− mice between 6 and 24 weeks was in the olfactory bulb (factor of 1.3) the largest increase was again seen in the septum (factor of 2.3). However, in contrast to the control mouse substrains, the hypothalamus demonstrated one of the largest increases in copper with development in IRP2 knockouts (factor of 2.1).
As we did not find a difference in copper between the control substrains at 6 weeks they were combined for further analysis (C57BL/6 controls). Compared with C57BL/6 controls, IRP2−/− mice had significantly reduced copper levels at 6 weeks in the parietal cortex, hippocampus, ventral striatum, thalamus, hypothalamus and whole brain (Figure 2, data for the parietal cortex and ventral striatum not shown). However, this brain region specific reduction in copper disappeared with development, with copper levels being equal between knockout and C57/BL6 control mice at 12 weeks. No difference was seen in the remaining brain regions at any age (Figure 2, showing only olfactory bulb and entorhinal cortex).
Figure 2.
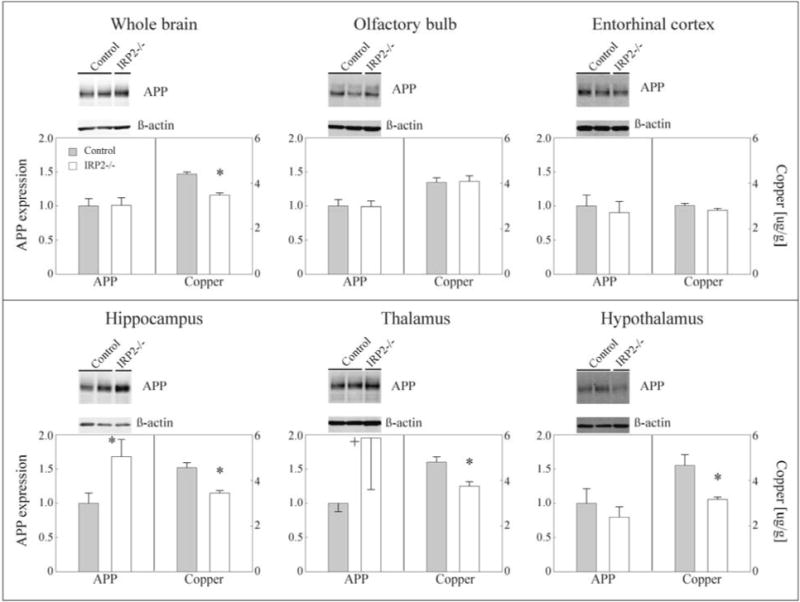
Copper levels and AβPP expression in IRP2−/− and C57BL/6 control mouse brain at 6 weeks of age (no difference between IRP2−/− and C57BL/6 control mice was seen for copper and AβPP expression at 24 weeks; data not shown). Gray bar: combined controls C57BL6/NTac + C57BL6/Jax, white bar: IRP2−/−. Left bar graphs show AβPP expression with corresponding left abscissa scale representing AβPP band intensity relative to β-actin. Representative western blot images above each graph (n = 5–7 per brain region and mouse strain; whole brain: n = 2–4). Right bar graphs show total copper levels with corresponding with right abscissa scale representing μg of copper/g wet tissue weight (n = 6–9 per brain region, mouse strain and age; whole brain: n = 4–8). Errors represent SEM; *p<0.05 IRP2−/− versus control, +p<0.1 IRP2−/− versus control.
Homology between mouse and human 5′-UTR of AβPP mRNA
To investigate the potential role of AβPP in the observed decrease in copper levels in young IRP2−/− mice we first compared the mRNA region previously identified as containing an iron response element in human AβPP with the respective sequence in mouse (Figure 3). Both sequences share 84% identity across this region, while the two homology clusters previously identified between human AβPP and human h-ferritin by Rogers et al. share 88% and 77% homology between human and mouse AβPP [15]. This is evidence that the same IRE like secondary mRNA structure predicted to form in the human AβPP mRNA may form in the mouse AβPP mRNA as well. Furthermore, while sequence identity for AβPP and h-ferritin in both species combined is only at 40% for the whole region, both homology clusters show a slightly increased sequence identity of 63% and 54%.
Figure 3.
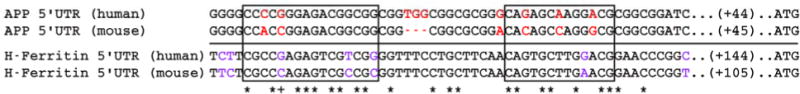
Iron response element sequence homology between human and mouse 5′-UTR mRNA of AβPP and h-ferritin. Sequence identity between human and mouse AβPP 5′-UTR mRNA is 84%. Differing nucleotides are shown in red. The comparable region containing the IRE in h-ferritin is shown below the line with differing nucleotides between human and mouse h-ferritin shown in purple. Consensus between all four sequences is marked with an asterix (*), while a plus sign (+) shows consensus only within species. Sequence identity between all four sequences is 40%. Boxed regions contain the two homology clusters between human AβPP and human h-ferritin described by Rogers et al. [15]. Sequence identity between mouse and human AβPP for these regions is 88% and 77% respectively, while identity between all four sequences is 63% and 54%.
AβPP expression in IRP2−/− and C57BL/6 control mice
After confirming a high percentage of sequence identity in the IRE regions of human and mouse AβPP 5′UTR mRNA we investigated the expression of AβPP in five brain regions as well as whole brain in knockout and C57BL/6 control mice. We chose to focus on the ages of 6 and 24 weeks since IRP2−/− mouse brain copper levels were reduced at 6 weeks but not different in any brain region at 24 weeks. Olfactory bulb and entorhinal cortex were included as control regions because no differences in copper levels between IRP2−/− and C57BL/6 control mice were seen in these regions at any age, thus leading us to expect no difference in AβPP expression either. Of the brain regions that showed a reduction in copper in 6 week old IRP2−/− mice we included the hippocampus, thalamus, hypothalamus and whole brain because the observed differences were more pronounced.
As expected, AβPP expression was equal between knockout and C57BL/6 control mice in the olfactory bulb and entorhinal cortex at 6 and 24 weeks (Figure 2, data not shown for 24 weeks). Furthermore, no difference in AβPP expression was seen in the hypothalamus or whole brain at either age. However, AβPP expression was significantly increased in knockouts in 6 week old hippocampus (p<0.05), as well as showing a trend towards increased expression in the thalamus (p<0.1). This increase in expression disappeared by 24 weeks in both regions (data not shown).
DISCUSSION
It is well known that copper and iron metabolism are linked; however, this relationship has been studied primarily in the context of copper regulating iron homeostasis. Here, we propose for the first time a mechanism by which iron may regulate copper levels in the brain – mediated by IRP2 and AβPP. To support this hypothesis we have compared copper levels and AβPP expression in several brain regions of mice with a targeted deletion of IRP2. We were able to show a significant reduction in copper for a number of different brain regions in knockout animals at 6 weeks of age. This corresponds with LaVaute, et al. who demonstrated that the IRP2 gene knock-out caused dysfunctional iron management in discreet brain regions, rather than producing ubiquitous deficits [23].
At this point it is not clear why copper levels equalize between IRP2−/− and control mice after six weeks of age. A possible explanation is the increased compensatory action of IRP1 with development. It has previously been shown that IRP1 is selectively activated by oxidative stress [28]. It has further been reported that the IRP2−/− mouse strain we have been working with in our study shows signs of neurodegeneration and iron accumulation in the brain after six months of age [23], although the extent of neurodegeneration is currently under debate [29,30]. It is therefore conceivable that oxidative stress increases with development in IRP2−/− mouse brains, which would then increase IRP1 activity. This is supported by the observation by Smith et al [31]. that IRP1+/− IRP2−/− mice show a much more severe phenotype than IRP2−/− mice and IRP1−/− IRP2−/− double knockout embryos do not even survive gestation. In contrast, Meyron-Holtz et al [13]. didn’t find an increase in IRP1 activity in IRP2−/− mice. Interestingly, in a previous study using the same brain samples we have studied here, we found loosely-bound iron to be decreased in IRP2−/− mouse brains at 6 weeks but equal with C57BL/6 controls in almost every brain region by 12 weeks, while non-heme iron was reduced throughout the whole age range studied (6 – 24 weeks) in several IRP2−/− mouse brain regions [24]. Therefore, the proposed compensatory action of IRP1 in the face of IRP2 loss needs to be addressed conclusively in a future study.
We found our results to be comparable to previous studies that measured brain copper concentrations. Ono et al. have reported whole brain copper concentrations of 8 week old C57BL/6J mice to be at 4.34 ± 1.3 μg/g, which is almost exactly what we found for 6 week old C57BL/6J mice (4.38 ± 0.1 μg/g) [32]. One difference was that our data showed the cerebellum to have the highest concentration of copper in young mice, while Ono et al. found the highest concentration in the hippocampus. Another study investigating brain copper levels in the mouse was done by Prohaska and Lukasewycz, who found slightly lower copper concentrations for whole brain (3.59 ± 0.18 μg/g) for 7 week old C57BL mice [33]. However, it is unclear which specific mouse substrain was used in this study. In a recent article by Jones et al. hippocampal levels of iron, copper and zinc were compared between 28 recombinant inbred mouse strains and their parental strains [34]. The authors observed a direct correlation between iron and copper levels, whereas we hypothesize that high iron levels can lead to lower levels of copper. However, as Jones et al. have indicated, the levels of iron and copper observed in these mice are probably not pathologically high or low and one can assume that metal homeostasis is not out of balance. In situations where this homeostasis is perturbed, as is the case in Alzheimer’s disease, the influence of iron on copper homeostasis may tip the scales toward increased copper export. This is simulated in our study by artificially lowering IRP2 to pathological levels.
To our knowledge this is the first time that a key protein in iron homeostasis has been implicated in the regulation of brain copper. This has particularly important implications for Alzheimer’s disease, wherein the homeostasis of both copper and iron has been shown to be dysfunctional. While amyloid plaques contain copper, iron and zinc in high concentrations [35,36], brain tissue copper levels are reduced in AD [37–39]. In contrast, brain tissue and neuronal iron levels are increased in AD [37,40–48], although some studies were unable to replicate this observation [38,49,50]. Here, we propose a mechanism by which the increase of iron and decrease of copper seen in AD brains may be linked. As both iron and copper can catalyze the generation of ROS it may be favorable for the brain to downregulate the level of one if the other is abnormally abundant, thus minimizing ROS production.
We postulate that this effect is mediated by the iron dependent degradation of IRP2. Very few studies have investigated IRP2 in Alzheimer’s disease. In a small study of 6 AD brains 2 were found to have an unusually stable IRE/IRP complex [51]. Furthermore, Smith et al. found IRP2 to be increased and associated with neurofibrillary tangles and neuropil threads in AD brains [52], a finding we were unable to reproduce [38]. Since AD is a disease developing slowly over the course of many years we expect protein expression changes to be very subtle with dysregulatory effects adding up over time. Therefore, further studies using sensitive techniques to measure protein abundance will have to establish the level of IRP2 in AD brain, as previous studies have only evaluated IRP2 expression using immunohistochemistry [38,52].
We expect the action of IRP2 on copper levels to be indirect; therefore we investigated intermediate molecules which might mediate the effect. One of the key molecules in the pathology of AD, AβPP, has been shown to have two copper binding sites, one in the Aβ sequence (CuBD-2) and the other in the N-terminal region (CuBD-1) [3–5]. CuBD-1 resembles copper binding domains of other known copper chaperones such as CCS and Atx1 [6] and is highly conserved between species [53]. Moreover, overexpression of AβPP causes a significant decrease in brain copper, whereas AβPP knockout mice have higher levels of copper in the brain, which seems to implicate AβPP in brain copper metabolism [8–12]. In vitro studies have further shown that human AβPP has a functional IRE at the 5′-UTR of its mRNA [15], indicating the involvement of iron regulatory proteins in the regulation of AβPP. We therefore assessed the role of AβPP as a possible mediator of the effect of IRP2 on brain copper levels by measuring the expression of AβPP in selected brain regions where copper was reduced in IRP2−/− mice. We were able to show a significant increase in AβPP expression in 6 week old hippocampus as well as a trend towards increased expression in the thalamus of IRP2−/− mice, which corresponds to a decrease in copper in both regions. However, no difference in AβPP expression was seen in the hypothalamus and whole brain although copper levels decreased in both. One possible explanation may be the specific type of IRE found in AβPP mRNA, which is different in sequence as well as secondary structure from the general consensus IRE [15,54]. This is further augmented by the fact that the regulation of the biosynthesis of ferritin, which has a typical IRE, is very rapid (4 h or less) [55], whereas iron dependent changes in AβPP expression have been observed only after 24 – 48 h [15,16]. This implicates the involvement of further mechanisms in the iron dependent regulation of AβPP expression, which could be differently regulated in different brain regions.
One potential influence on AβPP expression that we cannot exclude in the current scenario is the reported downregulation of AβPP expression by reduced copper itself via the AβPP promoter [56]. Bellingham et al. were able to show that decreasing intracellular copper leads to a decrease in AβPP expression in human fibroblasts, whereas increasing copper does not change AβPP expression. They went on to show that this effect is dependent on gene expression regulation via a 5′-AβPP promoter. We think that our study and this study by Bellingham et al. are looking at two different mechanisms. According to our hypothesis AβPP expression is upregulated by the loss of function of IRP2, which in turn leads to a decrease in copper. Bellingham, on the other hand, lowered the level of intracellular copper and then measured the effect on AβPP. In their discussion they state that this in fact supports the role of AβPP in copper homeostasis because a reduced level of copper requires less AβPP for further copper efflux – hence, the downregulation of AβPP. Therefore, one could argue that both mechanisms, the regulation of AβPP expression by copper levels and the regulation of copper levels via AβPP, coexist. Further studies will have to establish and characterize the impact of this interaction between both mechanisms in more detail.
We conclude then that IRP2 affects brain copper levels and that this may be mediated at least in part through AβPP. However, alterations in AβPP levels alone are insufficient to explain the entire effect of IRP2 on brain copper. Furthermore, it has yet to be elucidated why the reduction in copper is only seen in young IRP2−/− mice. The relationship between copper and iron in the brain is likely considerably more complex and additional pathways must be explored to completely characterize this relationship.
Acknowledgments
We want to thank Dr. RL Levine and Dr. JR Prohaska for suggestions on the manuscript as well as Waheed Baqai for suggestions on the statistical evaluation of results. This work was supported by the National Institute on Aging, NIH, AG20948.
References
- 1.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 2.Hamza I, Schaefer M, Klomp LW, Gitlin JD. Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc Natl Acad Sci U S A. 1999;96:13363–8. doi: 10.1073/pnas.96.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hesse L, Beher D, Masters CL, Multhaup G. The beta A4 amyloid precursor protein binding to copper. FEBS Lett. 1994;349:109–116. doi: 10.1016/0014-5793(94)00658-x. [DOI] [PubMed] [Google Scholar]
- 4.Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, Masters CL, Beyreuther K. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I) Science. 1996;271:1406–1409. doi: 10.1126/science.271.5254.1406. [DOI] [PubMed] [Google Scholar]
- 5.Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI. Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J Neurochem. 2000;75:1219–1233. doi: 10.1046/j.1471-4159.2000.0751219.x. [DOI] [PubMed] [Google Scholar]
- 6.Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem. 2003;278:17401–17407. doi: 10.1074/jbc.M300629200. [DOI] [PubMed] [Google Scholar]
- 7.Bayer TA, Multhaup G. Involvement of amyloid beta precursor protein (AbetaPP) modulated copper homeostasis in Alzheimer’s disease. J Alzheimers Dis. 2005;8:201–206. doi: 10.3233/jad-2005-8212. [DOI] [PubMed] [Google Scholar]
- 8.White AR, Reyes R, Mercer JF, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999;842:439–444. doi: 10.1016/s0006-8993(99)01861-2. [DOI] [PubMed] [Google Scholar]
- 9.Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, Masters CL, Bush AI, Li QX. Overexpression of Alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277:44670–44676. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- 10.Phinney AL, Drisaldi B, Schmidt SD, Lugowski S, Coronado V, Liang Y, Horne P, Yang J, Sekoulidis J, Coomaraswamy J, Chishti MA, Cox DW, Mathews PM, Nixon RA, Carlson GA, St George-Hyslop P, Westaway D. In vivo reduction of amyloid-beta by a mutant copper transporter. Proc Natl Acad Sci USA. 2003;100:14193–14198. doi: 10.1073/pnas.2332851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayer TA, Schafer S, Simons A, Kemmling A, Kamer T, Tepest R, Eckert A, Schussel K, Eikenberg O, Sturchler-Pierrat C, Abramowski D, Staufenbiel M, Multhaup G. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc Natl Acad Sci USA. 2003;100:14187–14192. doi: 10.1073/pnas.2332818100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellingham SA, Ciccotosto GD, Needham BE, Fodero LR, White AR, Masters CL, Cappai R, Camakaris J. Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J Neurochem. 2004;91:423–428. doi: 10.1111/j.1471-4159.2004.02731.x. [DOI] [PubMed] [Google Scholar]
- 13.Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, Rouault TA. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004;23:386–395. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Recalcati S, Alberghini A, Campanella A, Gianelli U, De CE, Conte D, Cairo G. Iron regulatory proteins 1 and 2 in human monocytes, macrophages and duodenum: expression and regulation in hereditary hemochromatosis and iron deficiency. Haematologica. 2006;91:303–310. [PubMed] [Google Scholar]
- 15.Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 16.Reznichenko L, Amit T, Zheng H, Vramovich-Tirosh Y, Youdim MB, Weinreb O, Mandel S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by ⊖-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer’s disease. J Neurochem. 2006;97:527–536. doi: 10.1111/j.1471-4159.2006.03770.x. [DOI] [PubMed] [Google Scholar]
- 17.Hart E, Steenbock H, Waddell J, Elvehjem C. Iron in nutrition. VII. Copper as a supplement to iron for hemoglobin building in the rat. J Biol Chem. 1928;77:797–833. [PubMed] [Google Scholar]
- 18.Millon E. De la presence normale de plusiers metaux dans le sang de l’homme, et de l’analyse des sels fixes contenus dans ce liquide. Comptes Rend de l’Acad des Sci a Paris. 1848:41–43. [Google Scholar]
- 19.Fox PL. The copper-iron chronicles: the story of an intimate relationship. Biometals. 2003;16:9–40. doi: 10.1023/a:1020799512190. [DOI] [PubMed] [Google Scholar]
- 20.Warburg O, Krebs H. Ueber locker gebundenes Kupfer und Eisen im Blutserum. Biochem Z. 1927:143–149. [Google Scholar]
- 21.Iwańska S, Strusińska D. Copper metabolism in different states of erythropoiesis activity. Acta Physiol Pol. 1978;29:465–74. [PubMed] [Google Scholar]
- 22.Collins JF. Gene chip analyses reveal differential genetic responses to iron deficiency in rat duodenum and jejunum. Biol Res. 2006;39:25–37. [PMC free article] [PubMed] [Google Scholar]
- 23.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 24.Magaki S, Mueller C, Yellon SM, Fox J, Kim J, Snissarenko E, Chin V, Ghosh MC, Kirsch WM. Regional dissection and determination of loosely bound and non-heme iron in the developing mouse brain. Brain Res. 2007;1158:144–150. doi: 10.1016/j.brainres.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paxinos G, Franklin KB. The Mouse Brain in Stereotaxic Coordinates: Compact. (Second) 2002:120. [Google Scholar]
- 26.Levites Y, Amit T, Mandel S, Youdim MB. Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol ⊖-epigallocatechin-3-gallate. FASEB J. 2003;17:952–954. doi: 10.1096/fj.02-0881fje. [DOI] [PubMed] [Google Scholar]
- 27.Hilbich C, Mönning U, Grund C, Masters CL, Beyreuther K. Amyloid-like properties of peptides flanking the epitope of amyloid precursor protein-specific monoclonal antibody 22C11. J Biol Chem. 1993;268:26571–7. [PubMed] [Google Scholar]
- 28.Pantopoulos K, Hentze MW. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc Natl Acad Sci U S A. 1998;95:10559–63. doi: 10.1073/pnas.95.18.10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galy B, Hoelter SM, Klopstock T, Ferring D, Becker L, Kaden S, Wurst W, Groene H, Hentze MW. Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse. Nat Genet. 2006;38:967–9. doi: 10.1038/ng0906-967. discussion 969–70. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh MC, Ollivierre-Wilson H, Cooperman S, Rouault TA. Reply to “Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse”. Nat Genet. 2006;38:969–970. doi: 10.1038/ng0906-967. [DOI] [PubMed] [Google Scholar]
- 31.Smith SR, Cooperman S, Lavaute T, Tresser N, Ghosh M, Meyron-Holtz E, Land W, Ollivierre H, Jortner B, Switzer R, Messing A, Rouault TA. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann N Y Acad Sci. 2004;1012:65–83. doi: 10.1196/annals.1306.006. [DOI] [PubMed] [Google Scholar]
- 32.Ono S, Koropatnick DJ, Cherian MG. Regional brain distribution of metallothionein, zinc and copper in toxic milk mutant and transgenic mice. Toxicology. 1997;124:1–10. doi: 10.1016/s0300-483x(97)00133-9. [DOI] [PubMed] [Google Scholar]
- 33.Prohaska JR, Lukasewycz OA. Effects of copper deficiency on the immune system. Adv Exp Med Biol. 1990;262:123–43. doi: 10.1007/978-1-4613-0553-8_11. [DOI] [PubMed] [Google Scholar]
- 34.Jones LC, Beard JL, Jones BC. Genetic analysis reveals polygenic influences on iron, copper, and zinc in mouse hippocampus with neurobiological implications. Hippocampus. 2008;18:398–410. doi: 10.1002/hipo.20399. [DOI] [PubMed] [Google Scholar]
- 35.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 36.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 37.Deibel MA, Ehmann WD, Markesbery WR. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: possible relation to oxidative stress. J Neurol Sci. 1996;143:137–142. doi: 10.1016/s0022-510x(96)00203-1. [DOI] [PubMed] [Google Scholar]
- 38.Magaki S, Raghavan R, Mueller C, Oberg KC, Vinters HV, Kirsch WM. Iron, copper, and iron regulatory protein 2 in Alzheimer’s disease and related dementias. Neurosci Lett. 2007;418:72–76. doi: 10.1016/j.neulet.2007.02.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plantin, Lying-Tunell, Kristensson Trace elements in the human central nervous system studied with neutron activation analysis. Biological Trace Element Research. 1987;13:69–75. doi: 10.1007/BF02796622. [DOI] [PubMed] [Google Scholar]
- 40.Thompson CM, Markesbery WR, Ehmann WD, Mao YX, Vance DE. Regional brain trace-element studies in Alzheimer’s disease. Neurotoxicology. 1988;9:1–7. [PubMed] [Google Scholar]
- 41.Dedman DJ, Treffry A, Candy JM, Taylor GA, Morris CM, Bloxham CA, Perry RH, Edwardson JA, Harrison PM. Iron and aluminium in relation to brain ferritin in normal individuals and Alzheimer’s-disease and chronic renal-dialysis patients. Biochem J. 1992;287(Pt 2):509–514. doi: 10.1042/bj2870509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samudralwar DL, Diprete CC, Ni BF, Ehmann WD, Markesbery WR. Elemental imbalances in the olfactory pathway in Alzheimer’s disease. J Neurol Sci. 1995;130:139–145. doi: 10.1016/0022-510x(95)00018-w. [DOI] [PubMed] [Google Scholar]
- 43.Loeffler DA, Connor JR, Juneau PL, Snyder BS, Kanaley L, DeMaggio AJ, Nguyen H, Brickman CM, LeWitt PA. Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J Neurochem. 1995;65:710–24. doi: 10.1046/j.1471-4159.1995.65020710.x. [DOI] [PubMed] [Google Scholar]
- 44.Kala SV, Hasinoff BB, Richardson JS. Brain samples from Alzheimer’s patients have elevated levels of loosely bound iron. Int J Neurosci. 1996;86:263–269. doi: 10.3109/00207459608986717. [DOI] [PubMed] [Google Scholar]
- 45.LeVine SM. Iron deposits in multiple sclerosis and Alzheimer’s disease brains. Brain Res. 1997;760:298–303. doi: 10.1016/s0006-8993(97)00470-8. [DOI] [PubMed] [Google Scholar]
- 46.Hautot D, Pankhurst QA, Khan N, Dobson J. Preliminary evaluation of nanoscale biogenic magnetite in Alzheimer’s disease brain tissue. Proc Biol Sci. 2003;270(Suppl 1):S62–S64. doi: 10.1098/rsbl.2003.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collingwood JF, Mikhaylova A, Davidson M, Batich C, Streit WJ, Terry J, Dobson J. In situ characterization and mapping of iron compounds in Alzheimer’s disease tissue. J Alzheimers Dis. 2005;7:267–272. doi: 10.3233/jad-2005-7401. [DOI] [PubMed] [Google Scholar]
- 48.Honda K, Smith MA, Zhu X, Baus D, Merrick WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, Liu Q, Moreira PI, Miller FP, Nunomura A, Shimohama S, Perry G. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem. 2005;280:20978–20986. doi: 10.1074/jbc.M500526200. [DOI] [PubMed] [Google Scholar]
- 49.Hallgren B, Sourander P. The non-haemin iron in the cerebral cortex in Alzheimer’s disease. J Neurochem. 1960;5:307–310. doi: 10.1111/j.1471-4159.1960.tb13369.x. [DOI] [PubMed] [Google Scholar]
- 50.Religa D, Strozyk D, Cherny RA, Volitakis I, Haroutunian V, Winblad B, Naslund J, Bush AI. Elevated cortical zinc in Alzheimer disease. Neurology. 2006;67:69–75. doi: 10.1212/01.wnl.0000223644.08653.b5. [DOI] [PubMed] [Google Scholar]
- 51.Pinero DJ, Hu J, Connor JR. Alterations in the interaction between iron regulatory proteins and their iron responsive element in normal and Alzheimer’s diseased brains. Cell Mol Biol. 2000;46:761–776. [PubMed] [Google Scholar]
- 52.Smith MA, Wehr K, Harris PL, Siedlak SL, Connor JR, Perry G. Abnormal localization of iron regulatory protein in Alzheimer’s disease. Brain Res. 1998;788:232–236. doi: 10.1016/s0006-8993(98)00002-x. [DOI] [PubMed] [Google Scholar]
- 53.Kong G, Miles L, Crespi G, Morton C, Ng H, Barnham K, McKinstry W, Cappai R, Parker M. Copper binding to the Alzheimer’s disease amyloid precursor protein. Eur Biophys J. 2008;37:269–279. doi: 10.1007/s00249-007-0234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hentze MW, Caughman SW, Casey JL, Koeller DM, Rouault TA, Harford JB, Klausner RD. A model for the structure and functions of iron-responsive elements. Gene. 1988;72:201–8. doi: 10.1016/0378-1119(88)90145-x. [DOI] [PubMed] [Google Scholar]
- 55.Rouault TA, Hentze MW, Dancis A, Caughman W, Harford JB, Klausner RD. Influence of altered transcription on the translational control of human ferritin expression. Proc Natl Acad Sci U S A. 1987;84:6335–9. doi: 10.1073/pnas.84.18.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bellingham SA, Lahiri DK, Maloney B, Fontaine SL, Multhaup G, Camakaris J. Copper depletion down-regulates expression of the Alzheimer’s disease amyloid-beta precursor protein gene. J Biol Chem. 2004;279:20378–20386. doi: 10.1074/jbc.M400805200. [DOI] [PubMed] [Google Scholar]