

ABSTRACT
Hantaviruses cause hemorrhagic fever with renal syndrome (HFRS) in the Old World and a highly fatal hantavirus cardiopulmonary syndrome (HCPS) in the New World. No vaccines or antiviral therapies are currently available to prevent or treat hantavirus disease, and gaps in our understanding of how hantaviruses enter cells challenge the search for therapeutics. We performed a haploid genetic screen in human cells to identify host factors required for entry by Andes virus, a highly virulent New World hantavirus. We found that multiple genes involved in cholesterol sensing, regulation, and biosynthesis, including key components of the sterol response element-binding protein (SREBP) pathway, are critical for Andes virus entry. Genetic or pharmacological disruption of the membrane-bound transcription factor peptidase/site-1 protease (MBTPS1/S1P), an SREBP control element, dramatically reduced infection by virulent hantaviruses of both the Old World and New World clades but not by rhabdoviruses or alphaviruses, indicating that this pathway is broadly, but selectively, required by hantaviruses. These results could be fully explained as arising from the modest depletion of cellular membrane cholesterol that accompanied S1P disruption. Mechanistic studies of cells and with protein-free liposomes suggested that high levels of cholesterol are specifically needed for hantavirus membrane fusion. Taken together, our results indicate that the profound dependence on target membrane cholesterol is a fundamental, and unusual, biophysical property of hantavirus glycoprotein-membrane interactions during entry.
IMPORTANCE
Although hantaviruses cause important human diseases worldwide, no specific antiviral treatments are available. One of the major obstacles to the development of new therapies is a lack of understanding of how hantaviruses hijack our own host factors to enter cells. Here, we identified multiple cellular genes that control the levels of cholesterol in cellular membranes to be important for hantavirus entry. Our findings suggest that high concentrations of cholesterol in cellular membranes are required at a specific step in the entry process—fusion between viral and cellular membranes—that allows escape of the hantavirus genome into the host cell cytoplasm to initiate infection. Our findings uncover a fundamental feature of the hantavirus infection mechanism and point to cholesterol-lowering drugs as a potential new treatment of hantaviral infections.
INTRODUCTION
Hantaviruses, members of the family Bunyaviridae of enveloped negative-strand RNA viruses, are associated with two distinct zoonotic disease syndromes in humans: hemorrhagic fever with renal syndrome (HFRS) and hantavirus cardiopulmonary/pulmonary syndrome (HCPS/HPS) (1–3). HFRS, caused by Old World hantaviruses (e.g., Dobrava virus, Puumala virus, and Hantaan virus [HTNV]) is widespread in Eastern Europe and Asia, with a global incidence of up to 200,000 cases per year (4). HCPS, caused by genetically distinct New World hantaviruses (e.g., Sin Nombre virus [SNV] and Andes virus [ANDV]), is endemic in the southwestern United States and in South America (5). Recently, an outbreak of HCPS in Yosemite National Park (California, USA) placed thousands of visitors from all over the world at risk of infection (6).
Hantaviruses are tri-segmented negative-strand RNA viruses (7). The large (L) and small (S) segments encode the viral-RNA-dependent RNA polymerase and the nucleoprotein (N), respectively, while the medium (M) segment encodes the envelope glycoprotein (8). The glycoprotein is translated as a single polypeptide, which is cotranslationally processed by the endoplasmic reticulum (ER)-resident signal peptidase to generate N-terminal (Gn) and C-terminal (Gc) subunits (9–11). Gn and Gc form heterodimeric oligomers embedded in the host-derived lipid bilayer of the virion and are necessary and sufficient to mediate viral entry into the cytoplasm of host cells (12–14). Pathogenic hantaviruses, such as ANDV and HTNV, and nonpathogenic hantaviruses, such as Prospect Hill virus (PHV), utilize β3 and β1 integrins, respectively, as entry receptors in vitro (15–17). Further, a glycosylphosphatidylinositol (GPI)-anchored protein, complement decay-accelerating factor (DAF/CD55) (18), and GC1QR (globular head of the complement C1q receptor) (19) have been implicated in hantavirus entry in cell culture. A recent study proposed roles for β2 integrin (CD18) heterodimers with CD11b (complement receptor 3 [CR3]) and CD11c (complement receptor 4 [CR4]) in HTNV entry and pathogenesis (20). However, the mechanistic roles of all of these host factors in hantavirus cell entry remain incompletely defined. Moreover, despite the identification of these host factors and their proposed implications for virulence, other host factors that influence hantavirus host range, tissue tropism, and pathogenesis likely await discovery.
To identify human genes required for ANDV entry and infection, we performed a genome-wide loss-of-function genetic screen in haploid human cells. While this work was in progress, Petersen et al. in 2014 published results from a similar screen and identified the host sterol regulatory element-binding protein (SREBP) pathway as a requirement for hantavirus entry (21). Our work confirms this finding and extends it by elucidating the mechanistic basis of the SREBP signaling requirement in hantavirus entry. We show that hantavirus membrane fusion and cytoplasmic escape are specifically and exquisitely sensitive to relatively small reductions in cellular membrane cholesterol that accompany disruption of the SREBP regulatory circuit. Studies with cells and purified liposomes reveal that this profound dependence on target membrane cholesterol is a fundamental, and unusual, biophysical property of hantavirus glycoprotein-membrane interaction during entry.
RESULTS
Multiple genes involved in cholesterol regulation are required for Andes virus entry.
To study hantavirus entry in a biosafety level 2 (BSL-2) setting, we engineered a recombinant vesicular stomatitis virus (rVSV) in which the VSV glycoprotein (G) was replaced with that of Andes virus (ANDV), a prototypic New World hantavirus. Using this agent (rVSV-ANDV GP), we performed a loss-of-function genetic screen in haploid human (HAP1) cells, as described previously (22–25). The screen identified seven genes that regulate cellular cholesterol metabolism (Fig. 1A). Four of these genes are critical components of the SREBP (sterol regulatory element-binding protein) cholesterol regulatory pathway (Fig. 1B): (i) SREBF2 (sterol regulatory element-binding transcription factor 2, here termed SREBP2) with 929 disruptive gene trap insertions (see Materials and Methods); (ii) MBTPS1 (membrane-bound transcription factor peptidase site 1, here termed site 1 protease [S1P]) with 273 disruptive insertions; (iii) MBTPS2 (membrane-bound transcription factor peptidase site 2, here termed site 2 protease [S2P]) with 218 disruptive insertions; and (iv) SCAP (SREBP cleavage-activating protein) with 142 disruptive insertions. In addition, three more genes encoding enzymes with roles in cholesterol biosynthesis were also identified: (i) LSS (lanosterol synthase) with 55 disruptive insertions, (ii) SQLE (squalene epoxidase) with 29 disruptive insertions, and (iii) ACAT2 (acetyl coenzyme A [acetyl-CoA] acetyltransferase 2) with 32 disruptive insertions (Fig. 1B). Most of the gene trap insertions were located toward the 5′ end of each gene and enriched for sense orientation insertions, which are more likely to impair gene function due to orientation-dependent activity of the splice acceptor. These findings suggest that ANDV entry is dependent on cellular cholesterol homeostasis.
FIG 1 .

A haploid genetic screen reveals multiple genes of the SREBP cholesterol regulatory pathway required for Andes virus entry. (A) Gene trap insertions in genes that are required for Andes virus entry in human HAP1 cells. For every gene, sites of proviral integration are depicted for unselected control cells (top) and cells selected with rVSV-ANDV GP (bottom). P values correspond to the enrichment of disruptive insertions (sense orientation insertions and insertions into exonic regions) in the virus-selected population over levels in the control. Insertion sites in both populations are also scored for orientational bias; integrations in the same transcriptional orientation as the gene (sense) are shown in blue, while those in the opposite orientation (antisense) are depicted in red. All genes with the exception of ACAT2 also show a statistically significant (P ≤ 0.005) bias for sense orientation integrations in the rVSV-ANDV GP-selected cells. (B) Cellular SREBP cholesterol regulatory pathway. Genes identified as hits in the screen are labeled in red. Golgi, Golgi apparatus; PP, pyrophosphate.
S1P is required for cell entry and infection by prototypic New World and Old World hantaviruses.
The S1P and S2P proteases play crucial roles in the SREBP signaling cascade that is initiated in response to cellular cholesterol depletion (26–28). Stepwise cleavage of SREBP2 by these proteases in the Golgi complex liberates the N-terminal transcription factor domain of SREBP2, allowing it to translocate to the nucleus and increase the expression of genes involved in cholesterol uptake and biosynthesis (29, 30) (Fig. 1B). Conversely, the inactivation of S1P or S2P promotes cellular cholesterol depletion by downregulating both extrinsic and intrinsic pathways for cholesterol acquisition (27, 28). Because S1P is a central regulator of the SREBP pathway for which a well-characterized small-molecule inhibitor exists, we focused our mechanistic studies on this enzyme.
We disrupted the S1P-encoding gene, MBTPS1, in the human osteosarcoma U2OS cell line by clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-mediated genome editing (31). We obtained a single-cell clone (S1P-#1) in which both alleles bore deletions at the expected site (Fig. 2A, top). These deletions were predicted to frameshift the S1P open reading frame (ORF) at amino acid positions 401 (allele 1) and 394 (allele 2), generating truncated polypeptides of 428 (allele 1) and 400 (allele 2) amino acid residues that terminate at downstream in-frame stop codons. Because these truncation mutants lack both Ser 414, which forms a part of the catalytic triad of the S1P serine protease, and the transmembrane anchor (residues 999 to 1021), which positions S1P to cleave SREBP2, they are predicted to be inactive. Finally, PCR with primers specific to the guide RNA (gRNA) editing site amplified S1P cDNA from wild-type (WT) but not S1P-#1 cells (Fig. 2A, bottom left), providing evidence that no WT MBTPS1 alleles are present in the latter cells. Therefore, S1P-#1 is an S1P-null cell line. To rule out any off-target effects of the CRISPR/Cas9-mediated knockout, we reconstituted these cells with a Flag-tagged version of the S1P protein (Fig. 2B, bottom right). Both the S1P-null cells and the reconstituted cells resembled WT U2OS cells in morphology and growth characteristics (data not shown).
FIG 2 .

S1P is required for hantavirus infection. (A) The S1P gene was knocked out in U2OS cells by using the CRISPR/Cas9 system. (Top) WT S1P gene sequence aligned with the sequences of both of the alleles in the S1P knockout (S1P-#1) cell clone. The gRNA target sequence is marked in red. The protospacer-adjacent motif (PAM) sequence of the gRNA target site is underlined. (Lower left) RT-PCR results for WT and S1P knockout (S1P-#1) cells using gRNA target site-specific primers. Human NPC1-specific primers were used as a control. (Lower right) S1P protein expression by anti-Flag immunostaining in S1P-#1 cells stably expressing Flag-tagged S1P. (B) WT and S1P knockout (S1P-#1) and S1P-reconstituted (S1P knockout cells stably expressing Flag-tagged S1P) U2OS cells were exposed to the indicated rVSVs at a multiplicity of infection (MOI) of 0.1 to 0.2 IU per cell. Infected (eGFP-positive) cells were visualized by fluorescence microscopy at 12 to 16 h postinfection. (C, left) Infected cells from the experiment in panel B were enumerated and normalized to infection in WT cells (n = 6; two independent experiments). (Right) WT, S1P knockout, and S1P-reconstituted cells were exposed to authentic ANDV at an MOI of 0.1 to 0.2 PFU per cell. Infection was scored at 72 h postinfection by enumerating viral-antigen-positive cells and was normalized to infection in WT U2OS cells.
We next exposed WT and S1P-#1 U2OS cells to rVSVs encoding GPs from ANDV, Hantaan virus (HTNV), or VSV. ANDV GP- and HTNV GP-mediated infections were reduced >95% in the S1P-null cells and could be fully rescued by ectopic expression of the S1P cDNA. In contrast, VSV G-mediated infection was only modestly inhibited in the S1P-null cells (Fig. 2B and C). Crucially, authentic ANDV resembled rVSV-ANDV GP in its dependence on S1P for infection (Fig. 2C, right). Taken together, these results indicate that S1P is required for cell entry and infection by the prototypic New World and Old World hantaviruses, ANDV and HTNV.
A small-molecule inhibitor of S1P selectively inhibits cell entry and infection by hantaviruses.
To corroborate the role of S1P in hantavirus entry, we tested the effect of a specific small-molecule inhibitor of S1P, PF-429242 (herein termed the S1P inhibitor) (32, 33), on infection by rVSVs bearing hantavirus glycoproteins. S1P inhibitor treatment reduced infection by rVSV-ANDV GP and rVSV-HTNV GP in a dose-dependent manner in U2OS cells. In contrast, rVSV-G, which fuses in early endosomes (34), and rVSV-LASV GP (Lassa virus, an Old World arenavirus), which fuses in late endosomes (35), were only modestly affected, suggesting that S1P inhibitor does not cause gross perturbations in viral trafficking to early and late endosomal compartments (Fig. 3A).
FIG 3 .

A small-molecule inhibitor of S1P selectively inhibits hantavirus entry. (A) U2OS cells pretreated with the indicated concentrations of the S1P inhibitor PF-429242 or the 1% DMSO vehicle for 24 h were exposed to the indicated viruses (0.1 to 0.2 IU per cell) (n = 3; representative of 3 independent experiments). After 2 h of incubation, NH4Cl (20 mM, final concentration) was added to the medium to prevent subsequent rounds of infection. Infected cells were enumerated as described for Fig. 2. (B) HUVECs were pretreated with the indicated concentrations of the S1P inhibitor or the 1% DMSO vehicle for 24 h and then exposed to authentic hantaviruses (1 PFU per cell). Viral-antigen-positive cells were enumerated by 24 h postinfection.
To assess whether S1P is also required for hantavirus infection in a human cell type relevant to in vivo pathogenesis, we pretreated primary human umbilical vein endothelial cells (HUVECs) with the S1P inhibitor and exposed them to authentic hantaviruses (Fig. 3B). We found that the S1P inhibitor substantially inhibited infection by HTNV and Sin Nombre virus (SNV), a pathogenic New World hantavirus, at concentrations that caused little or no effect on cell viability or proliferation (see Fig. S1 in the supplemental material). These findings confirm that inactivation of S1P selectively impairs hantavirus entry and infection in human cells. They also set the stage for mechanistic studies using the S1P inhibitor to determine the precise role of the SREBP regulatory pathway in hantavirus entry.
S1P inactivation blocks hantavirus entry by depleting cellular membrane cholesterol.
We postulated that hantavirus entry requires S1P because its inactivation disrupts the SREBP2-dependent signaling circuit, thus leading to depletion of cellular cholesterol, as shown previously (26–28). To begin to test this hypothesis, we pretreated U2OS cells with the S1P inhibitor and incubated the live cells with a reporter for the plasma membrane/endosomal cholesterol pool: the cholesterol-binding domain of the Clostridium perfringens θ toxin fused to the enhanced blue fluorescent protein (eBFP2-θD4) (36). eBFP2-θD4 staining at the cell surface was dramatically reduced in S1P inhibitor-treated cells relative to that in dimethyl sulfoxide (DMSO)-treated controls but could be restored by brief (5-min) incubation with a cholesterol:2-hydroxypropyl-β-cyclodextrin complex (chol:CD) (Fig. 4A). Thus, cells lacking S1P activity suffered a reduction in plasma membrane and/or endosomal cholesterol levels, even though they were maintained in growth medium replete with cholesterol-rich low-density lipoproteins. A likely explanation for this apparent paradox is that interruption of the SREBP2 signaling pathway downregulates not only the biosynthesis of cholesterol but also its uptake (37, 38; see Discussion for details).
FIG 4 .
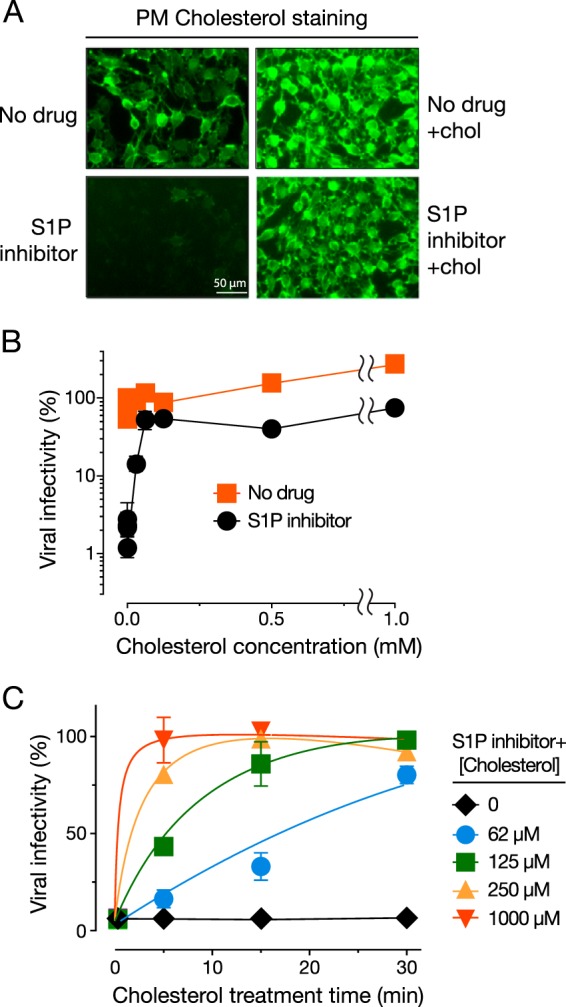
S1P inactivation blocks hantavirus entry by depleting the cellular plasma membrane/endosomal cholesterol pool. (A) U2OS cells were pretreated for 24 h with the S1P inhibitor (25 µM) or the 1% DMSO vehicle and then exposed to cholesterol:cyclodextrin (chol:CD) complexes (1 mM for cholesterol) for 5 min. Live cells were stained with eBFP2-θD4 (pseudocolored green) to mark the plasma membrane and/or endosomal cholesterol and imaged by fluorescence microscopy. (B) U2OS cells were pretreated with the S1P inhibitor as described above and then supplemented with various amounts of cholesterol by chol:CD treatment for 1 h. Cells were then exposed to rVSV-ANDV GP (0.1 IU per cell) (n = 3; results are representative of three independent experiments). Infected cells were enumerated at 12 to 16 h postinfection. (C) U2OS cells were pretreated with S1P inhibitor as described above and then exposed to different concentrations of the chol:CD complexes for the indicated lengths of time. Cells then underwent a single-cycle infection by rVSV-ANDV GP (1.5 IU per cell) and were scored for infection as described above (n = 4; two independent experiments).
To directly evaluate the requirement for membrane cholesterol in hantavirus entry, cells pretreated with the S1P inhibitor were incubated with increasing concentrations of chol:CD for 1 h to restore cholesterol levels and then exposed to rVSV-ANDV GP. Cholesterol repletion of S1P inhibitor-treated cells rescued viral entry in a concentration-dependent manner; the addition of as little as 62 µM cholesterol restored ANDV GP-mediated infection to essentially full levels (Fig. 4B). Additional time course experiments demonstrated that the effect of cholesterol loading on viral entry was also time dependent, with the highest tested dose of cholesterol (1 mM) effecting full rescue of infection in as little as 5 min (Fig. 4C). The rapid and complete rescue of infection effected by exogenous cholesterol in these experiments strongly suggests that hantaviruses have a profound and direct requirement for cellular membrane cholesterol during entry.
Cellular membrane cholesterol depletion does not affect viral attachment to cells.
We next sought to identify the step(s) in hantavirus entry that is strongly dependent on the levels of cellular membrane cholesterol. To determine if attachment of viral particles to the cell surface is affected by depletion of membrane cholesterol, we pretreated cells with the S1P inhibitor and then exposed them to VSV-ANDV GP particles in which the viral phosphoprotein, P, was fused to the fluorescent protein mNeonGreen [VSV(mNG-P)-ANDV GP] at 4°C (Fig. 5A) (see Materials and Methods for details). Cells were washed extensively in the cold, and flow cytometry was used to measure the fluorescence associated with cell surface-bound viral particles. S1P inhibitor treatment did not significantly diminish viral attachment to cells, suggesting that cholesterol depletion does not affect this entry step. Unexpectedly, however, incubation of cells with chol:CD prior to the addition of viral inoculum did slightly increase virus-cell attachment (data not shown, but see Fig. 5B).
FIG 5 .
Reduction in cellular membrane cholesterol does not affect viral attachment to cells. (A) Schematic of the virus attachment assay. (B) U2OS cells were treated with the S1P inhibitor (25 µM) or the 1% DMSO vehicle for 24 h and then with 62.5 µM cholesterol for the indicated amounts of time. To assess viral attachment, cells were exposed to fluorescein-labeled rVSV-ANDV GP at 4°C (1.5 IU per cell). Bound virus was quantified by flow cytometry. rVSV-ANDV GP binding and infection were performed in parallel, and values are normalized to binding and infection, respectively, in vehicle-treated cells (n = 4; two independent experiments).
To ensure that cholesterol-mediated restoration of rVSV-ANDV GP infection in cells treated with S1P inhibitor was not an artifact of increased viral attachment, we incubated inhibitor-treated cells with chol:CD (62.5 µM cholesterol) for 0 to 60 min and then performed viral attachment and infection assays in parallel. rVSV-ANDV GP attachment to cells increased modestly, reaching a plateau at 30 min, whereas infection increased dramatically (1,700) and continuously from 0 to 60 min (Fig. 5B). This disconnect between viral attachment and infectivity restoration upon cholesterol replenishment suggests that cholesterol depletion inhibits ANDV entry predominantly at a postattachment step.
Reduction in cellular membrane cholesterol delays, but does not block, viral internalization.
Entry by both ANDV and HTNV requires viral internalization into endosomes (39–41). To determine if cholesterol depletion inhibits viral internalization, we pretreated cells with the S1P inhibitor and exposed them to VSV(mNG-P)-ANDV GP particles at 4°C. Cells were then washed and shifted to 37°C to allow virus uptake. Cells harvested at each time point were incubated with trypsin to strip cell surface-bound viral particles (Fig. 6A), and internalized particles were detected by fluorescence microscopy (Fig. 6B) and flow cytometry (Fig. 6C and D). Viral particles were rapidly internalized in untreated cells. In contrast, little viral internalization was observed in S1P inhibitor-treated cells at 0 to 30 min postinfection. Nevertheless, these cells had internalized an amount of virus similar to that internalized by their untreated counterparts by 60 min at 37°C. In contrast, S1P inhibitor treatment (25 µM) did not significantly affect internalization of VSV(mNG-P) particles bearing the VSV glycoprotein (G) at any time point. A similar kinetic delay in internalization of viral particles containing ANDV GP, but not VSV G, was observed in S1P-null U2OS cells (Fig. S2). Thus, S1P inhibitor treatment slows, but does not block, viral internalization. We conclude, therefore, that cholesterol depletion acts predominantly at a step downstream of viral internalization.
FIG 6 .

Reduction in cellular membrane cholesterol delays, but does not block, viral internalization. (A) Schematic of the virus internalization assay. (B) U2OS cells were treated with the S1P inhibitor (25 µM) or the 1% DMSO vehicle for 24 h and exposed to VSV(mNG-P)-ANDV GP particles at 4°C. Following internalization of virus at 37°C for the indicated times (in minutes), remaining surface-bound virus particles were removed by a brief trypsin treatment. Cells were harvested and fixed with 4% PFA, and internalized virus was measured by microscopy (B) and flow cytometry (C and D) (n = 4; two independent experiments).
Cholesterol depletion inhibits ANDV GP-mediated virus-plasma membrane fusion.
We postulated that cholesterol depletion affects viral fusion with endosomal membranes and escape into the cytoplasm. To test this hypothesis, we established a “fusion infection” assay, in which viral fusion with the plasma membrane (and ensuing cytoplasmic release) is induced by extracellular acid pH. We found that ANDV GP-mediated fusion infection was triggered at a threshold pH between 5.8 and 6.1 but occurred optimally at pH 5.5 (see Fig. S3 in the supplemental material). Accordingly, cells were exposed to rVSV-ANDV GP at 4°C and then briefly incubated at 37°C, with the growth medium adjusted to pH 7.0 or pH 5.5. This medium was then replaced with standard growth medium (pH 7.0) containing NH4Cl to block viral membrane fusion via the normal endocytic route. We found that extracellular acid pH, but not neutral pH, could rapidly trigger viral entry into the cytoplasm (Fig. 7A), consistent with acid-dependent viral fusion with the plasma membrane. Similar levels of viral infectivity were obtained with the standard infection and fusion infection protocols carried out in parallel (Fig. 7A). Fusion infection was essentially complete at 1 min after exposure of virus-bound cells to acid pH (Fig. 7B). Thus, acid-induced fusion infection mediated by ANDV GP is both efficient and rapid.
FIG 7 .

Reduction in cellular membrane cholesterol blocks ANDV GP-mediated membrane fusion. (A) U2OS cells were exposed to rVSV-ANDV GP (2.2 IU per cell). For the infection experiment, 20 mM NH4Cl was added to the medium to allow only a single round of infection. For fusion infection, after the binding of virus to the cells at 4°C, cells were exposed to either pH 5.5 or 7.0 medium at 37°C for 1 min and then returned to medium containing 20 mM NH4Cl. Cells were incubated at 37°C overnight and scored for infection the next day (n = 4; two independent experiments). (B) A fusion infection experiment was performed on U2OS cells as described for panel A, with the pH treatment at 37°C for the indicated times (n = 2). (C) U2OS cells were treated with the S1P inhibitor (25 µM) or the 1% DMSO vehicle for 24 h and then supplemented with 1 mM cholesterol (as a chol:CD complex) for 5 min. rVSV-ANDV GP (2.2 IU per cell) was bound to the cells at 4°C, exposed to pH 5.5 or 7.0 medium for 1 min at 37°C, and then returned to medium containing 20 mM NH4Cl. Cells were fixed 16 h later and scored for virus infection (eGFP expression) (n = 4; two independent experiments). (D) Fusion infection using DiD-labeled rVSV-ANDV GP (2.6 IU per cell) was performed on U2OS cells as described for panel C except that the cells were fixed immediately after the low-pH treatment and imaged for their DiD dequenching signal. (E) Quantitative analysis of the DiD dequenching in panel D (see Materials and Methods for details; n = 4; two independent experiments).
We next pretreated cells with the S1P inhibitor and examined their capacity to support fusion infection by rVSV-ANDV GP. Inhibitor treatment reduced viral fusion infection by >90%, in a manner that could be fully reversed by cholesterol replenishment (Fig. 7C). In contrast, S1P inhibitor treatment had little effect on alphavirus fusion infection and reduced alphavirus infection more modestly than rVSV-ANDV GP infection via the endocytic route (Fig. S4). This result was expected from previous work (42, 43) which showed that alphaviruses specifically require cholesterol for membrane fusion, but only at low concentrations (also see Fig. 8B). These findings strongly suggest that cholesterol depletion interferes directly with hantavirus membrane fusion. They also indicate that the membrane fusion mechanism of hantaviruses is more profoundly sensitive to reductions in membrane cholesterol than that of alphaviruses.
FIG 8 .
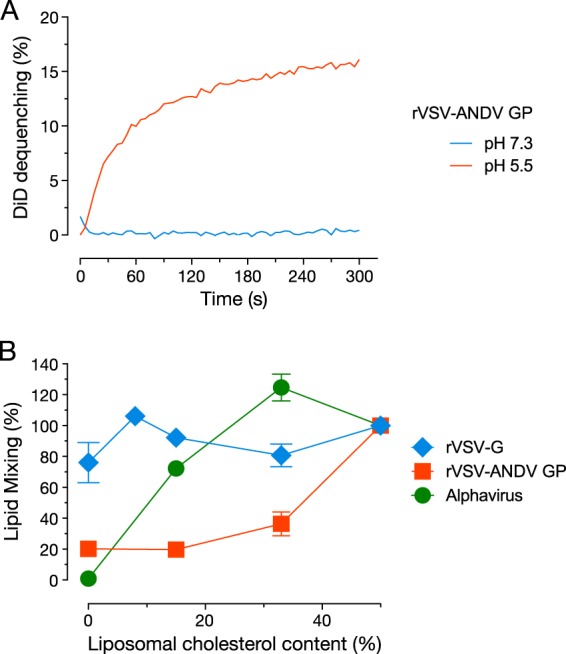
The cell-free-virus–liposome lipid mixing assay recapitulates the unusual cholesterol requirement for hantavirus membrane fusion. (A) DiD-labeled rVSV-ANDV GP (1 × 107 IU) was mixed with liposomes containg 50 mol% cholesterol at the indicated pH values. (B) DiD-labeled rVSV-G (2.8 × 106 IU), rVSV-ANDV GP (2.89 × 106 IU), and the alphavirus Semliki Forest virus (3.75 × 108 IU) were incubated with liposomes containing the indicated amounts of cholesterol at pH 5.5. The final extent of DiD dequenching was plotted (n = 4; two independent experiments).
Depletion in cellular membrane cholesterol inhibits hantavirus membrane fusion at or near the lipid mixing step and blocks cytoplasmic delivery of viral matrix protein.
Fusion between viral and cellular membranes is postulated to occur in a stepwise manner, with initial mixing of the outer membrane leaflets (hemifusion), followed by coalescence of the inner membrane leaflets to form a fusion pore, and expansion of this pore to allow passage of the viral core into the cytoplasm (44, 45). We sought to determine if cholesterol depletion inhibits hantavirus membrane fusion by abolishing the initial lipid mixing step or, instead, by acting at a later step in the fusion reaction. Accordingly, we developed a fluorescence dequenching assay to directly interrogate virus-plasma membrane lipid mixing. rVSV particles were labeled with self-quenching concentrations of the lipophilic fluorescent dye 4-chlorobenzenesulfonate (DiD), which intercalates into viral membranes and fluoresces upon dilution following membrane merger or detergent solubilization (Fig. S5). Cells were exposed to DiD-labeled rVSV-ANDV GP at 4°C and then shifted briefly to 37°C in neutral or acidic medium, as described above for the fusion infection assay. Incubation of virus-bound cells at acid pH, but not neutral pH, greatly increased the cell-associated DiD signal, as monitored by fluorescence microscopy (Fig. 7D and E), providing evidence for ANDV GP-mediated fusion between the viral lipid bilayer and the plasma membrane. We used the DiD dequenching assay to assess the consequence of S1P inhibitor treatment on virus-cell lipid mixing during fusion infection. Inhibitor treatment reduced lipid mixing almost to background levels (Fig. 7D and E). Strikingly, cholesterol replenishment not only restored lipid mixing but greatly enhanced it, even relative to that in untreated controls (Fig. 7D and E).
Finally, we used the intracellular distribution of the virion-associated VSV M (matrix) protein as a marker for productive endosomal membrane penetration and cytoplasmic escape, as described previously (22, 46). WT and S1P-null U2OS cells were exposed to rVSV-G or rVSV-ANDV GP in the presence of cycloheximide (to block new M protein synthesis) and then immunostained to visualize the incoming M protein (Fig. S6). Entry by either virus into WT cells caused the redistribution of M protein throughout the cytoplasm, as shown previously (22, 46). In contrast, perinuclear M puncta were extensively observed in S1P-null cells exposed to rVSV-ANDV GP but not rVSV-G, indicative of a membrane penetration block (Fig. S6). Taken together, these findings suggest that cellular cholesterol depletion induced by genetic or pharmacological disruption of the SREBP2 signaling circuit blocks hantavirus entry at the membrane fusion step, consequently preventing virus delivery into the cytoplasm.
Virus-liposome fusion mediated by ANDV GP is highly cholesterol dependent.
We considered two possible (nonexclusive) explanations for the preceding findings. First, ANDV GP may require high levels of cholesterol in the target membrane to carry out the fusion reaction. Alternatively, cholesterol depletion may affect the distribution and/or function of a cellular protein (e.g., a receptor) that is required for ANDV membrane fusion. To test the first hypothesis, we developed an in vitro assay for membrane fusion between viral particles and protein-free unilamellar membrane vesicles (liposomes). DiD-labeled rVSV-ANDV GP particles were incubated at neutral or acid pH with liposomes containing 50% cholesterol (mol/mol), and DiD dequenching concomitant with virus-liposome fusion was monitored by fluorimetry. A membrane fusion signal was detected at acid pH but not at neutral pH, as expected (Fig. 8A). Experiments with liposome preparations containing a range of cholesterol concentrations (Fig. 8B) revealed that ANDV GP-dependent virus-liposome fusion was strikingly dependent on target membrane cholesterol; lipid mixing was inhibited by 60% when cholesterol was lowered from 50% to 30% and fell to baseline levels when cholesterol was reduced further. In contrast, target membrane cholesterol was dispensable for VSV G-liposome fusion, which occurred efficiently under all conditions tested. Alphavirus fusion displayed a pattern dissimilar from those of ANDV and VSV; it was relatively unaffected at the higher cholesterol concentrations but was completely abolished in the absence of cholesterol, as shown previously (42, 43, 47) (Fig. 8B). Moreover, the different cholesterol dependencies of these three enveloped viruses for fusion with protein-free membranes were concordant with their sensitivity to S1P inhibition and cholesterol depletion in cell-based assays (Fig. 7). We conclude that the unusually strong dependence on target membrane cholesterol is a distinctive property of the hantavirus fusion mechanism that derives primarily from the interactions of its glycoproteins with host membranes and not host proteins.
DISCUSSION
Using a loss-of-function genetic screen in haploid human cells, we show that gene networks involved in the sensing, regulation, and biosynthesis of cellular cholesterol are required for hantavirus entry (Fig. 1A and B). We have implicated three genes that encode cholesterol-biosynthetic enzymes (LSS, SQLE, and ACAT2) (Fig. 1A), in addition to the four genes (SREBP2, S1P, S2P, and SCAP) that play key roles in the SREBP pathway, also previously identified by Petersen et al. (21). The identification of several components of these cholesterol pathways hinted strongly at a requirement for the metabolite cholesterol rather than for the individual gene products. Strikingly, these networks did not appear as hits in numerous published haploid genetic screens for host factors involved in viral infection and toxin-mediated cytotoxicity (22–25, 48). Results described previously and herein provide a likely explanation for this observation: hantavirus entry and infection appear to be unusually sensitive to the perturbation of cellular membrane cholesterol. ANDV GP-mediated entry of retrovirus particles and HTNV infection were previously shown to be sensitive to treatment of cells with methyl-β-cyclodextrin, which greatly depletes cellular cholesterol levels (18, 49). However, the mechanistic basis of these effects was not determined.
Genetic or pharmacological disruption of S1P activity in a human osteosarcoma cell line, as well as in primary human endothelial cells (HUVECs), strongly inhibited infection by hantaviruses with zoonotic potential belonging to both the Old World and New World clades (Fig. 2 and 3). This blockade could be fully overcome by brief supplementation of cells with cholesterol (Fig. 4). Independently of its role in cholesterol regulation, S1P enzymatic activity is required for proteolytic processing and maturation of glycoproteins from some bunyaviruses (e.g., Crimean-Congo hemorrhagic fever virus) (50) and arenaviruses (e.g., Lassa virus and lymphocytic choriomeningitis virus) (51, 52). However, our findings argue against such a direct role for S1P in hantavirus entry; the kinetics and extent of S1P inhibitor bypass obtained with cholesterol supplementation (Fig. 4) show that reductions in hantavirus entry and infection can be fully explained by the depletion of membrane cholesterol secondary to S1P loss.
Why is an intact cholesterol homeostatic network required for hantavirus entry when target cells have been cultured in serum-containing medium replete with cholesterol-laden low-density lipoproteins (LDL)? Several considerations may resolve this apparent paradox. First, because functional SREBP signaling is required for continued transcription of the LDL receptor gene (LDLR), disruption of this regulatory circuit is predicted to greatly reduce the levels of cell surface LDLR, thereby limiting cholesterol acquisition (37, 38). Moreover, because mutant cells cannot sense that they are becoming cholesterol depleted, they cannot respond by upregulating cholesterol biosynthesis. It is therefore expected that cells will become cholesterol deficient even “in midst of plenty.” Second, previous work offers a different possible explanation for why the biosynthetic genes SQLE, LSS, and ACAT2 hit in our screen. Sugii and coworkers (53) showed that Chinese hamster ovary (CHO) cells continue to synthesize cholesterol even when exogenous cholesterol is available in the form of LDL and suggested that loss of endogenous cholesterol synthesis selectively reduces endo/lysosomal cholesterol levels. Accordingly, we postulate that the disruption of cholesterol-biosynthetic genes inhibits acid-dependent hantavirus membrane fusion in endocytic compartments. Third, our findings, together with those of Petersen and coworkers (21), suggest that membrane cholesterol levels are only modestly reduced under these conditions. The cholesterol-binding domain of the Clostridium perfringens θ toxin, a probe that is especially sensitive to small perturbations in membrane cholesterol, reported a significant reduction in plasma membrane cholesterol in S1P inhibitor-treated cells (Fig. 4A), whereas a less sensitive probe, filipin III complex from Streptomyces filipinensis, did not (data not shown). Similarly, Petersen et al. measured an approximately 30% total cholesterol depletion in S1P inhibitor-treated cells (see Fig. S5 in reference 21). More-profound cholesterol depletion, typically associated with broad-spectrum viral inhibition, likely requires that cells be serum starved and/or treated with agents (e.g., cyclodextrins) that extract membrane cholesterol (54). These observations suggest that hantaviruses—more than most viruses and toxins—require high levels of cellular membrane cholesterol for optimal cell entry and infection.
How does modest cholesterol depletion attendant to S1P inhibitor treatment block hantavirus entry? Our results are in partial agreement with those of Petersen et al., who reported that these conditions did not affect ANDV GP-dependent viral attachment to cells but inhibited viral internalization into endosomes, the presumptive site(s) of hantavirus membrane fusion (21). In our hands, however, S1P inhibition slowed, but did not block, rVSV-ANDV GP internalization into endosomes. Instead, using assays for virus-membrane fusion, we provide evidence that cholesterol depletion selectively and profoundly inhibits GP-catalyzed fusion between viral and cellular membranes, thus blocking subsequent escape of viral cores into the cytoplasm (Fig. 7; see also Fig. S6 in the supplemental material). We therefore propose that S1P inhibition and attendant cellular cholesterol depletion act predominantly by blocking hantavirus membrane fusion, with smaller contributions from one or more upstream steps, including viral attachment and internalization.
Previous work has implicated multiple protein receptors in hantavirus infection and pathogenesis, but an understanding of their mechanistic roles in entry remains limited. Here, we show, for the first time, that ANDV GP can drive lipid mixing between viral and model membranes in a manner that requires acid pH but not cellular proteins (Fig. 8). This observation suggests that cellular receptors are not critically required to trigger at least some hantavirus membrane fusion machines (but see below).
Various target membrane cholesterol compositions in the virus-liposome assay allowed us to directly assess the effect of cholesterol on hantavirus membrane fusion. We found that ANDV GP required much higher levels of cholesterol for efficient lipid mixing than did the divergent glycoproteins from a rhabdovirus and an alphavirus (Fig. 8). These results closely mirrored the relative sensitivities of all three viral glycoproteins to cholesterol perturbation in cells, strongly suggesting that target membrane cholesterol is a biophysical requirement for hantavirus membrane fusion that is independent of cellular proteins.
We observed that the pH optimum for ANDV GP-mediated fusion with the plasma membrane is ~5.5 (Fig. S3), raising the possibility that ANDV penetrates from late endosomes. Interestingly, if this hypothesis is correct, our results from the liposome fusion assay (Fig. 8B) suggest that ANDV GP requires a higher concentration of cholesterol than might be available in late endosomes; the limiting membranes of these compartments are thought to contain less cholesterol than the plasma membrane (~35 mol%) (55–58). More work is thus needed (i) to identify the subcellular sites of hantavirus membrane fusion to assess whether cellular membrane constituents, including proteins (e.g., receptors) and lipids, “tune” the precise viral requirement for acid pH and membrane cholesterol and (ii) to determine if hantaviruses might exploit heterogeneities in the distribution of endosomal membrane cholesterol (59), such as cholesterol-rich microdomains, for fusion. The hantavirus-membrane fusion assays described herein may provide useful experimental systems to assess these hypotheses, as well as to identify additional cellular factors that modulate hantavirus glycoprotein fusogenicity, lipid mixing, and events downstream of the initial membrane merger, such as the formation of fusion pores.
Why do hantaviruses require high levels of target membrane cholesterol for fusion? Alphaviruses, which also require target membrane cholesterol, albeit at lower levels, provide one potentially applicable paradigm (43). Thus, like alphavirus E1, ANDV GP may need to bind specifically to cholesterol in order to insert into the target membrane. Previous work with synthetic peptides and liposomes corresponding to the putative ANDV fusion loop may support this hypothesis (60). Alternatively or in addition, it is conceivable that cholesterol modulates hantavirus membrane fusion via its effects on target membrane fluidity and rigidity (61, 62). These properties may directly affect the insertion of a viral fusion peptide(s) into the cellular lipid bilayer, the conformation and disposition of that membrane-inserted segment(s), and the propensity of the outer bilayer leaflet to deform prior to hemifusion with its viral counterpart. More studies are required to differentiate among these and other possible explanations for the unusual cholesterol dependence of hantavirus membrane fusion and to determine if this feature is shared by other bunyaviruses. Finally, it would be interesting to explore the possibility that cholesterol use by hantaviruses (or changes in that use) influences viral virulence or transmissibility.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney Vero cells and 293T human embryonic kidney cells were cultured at 37°C with 5% CO2 in high-glucose Dulbecco’s modified Eagle medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 1% GlutaMAX (Life Technologies), and 1% penicillin-streptomycin (Life Technologies). Human osteosarcoma U2OS cells were similarly maintained in modified McCoy’s 5A medium (Life Technologies) supplemented with the above-named reagents. HAP1 were cultured as described previously (22). Human umbilical vein endothelial cells (HUVECs; Lonza) were maintained in endothelial cell growth medium (EGM) supplemented with EGM-SingleQuots (Lonza) at 37°C with 5% CO2. A recombinant VSV bearing the VSV G glycoprotein was propagated on Vero cells, as described previously (67). HTNV strain 76-118 (63), SNV strain CC107 (64) and ANDV strain Chile-9717869 (65) were kindly provided by J. Hooper (USAMRIID) and propagated in Vero E6 cells as previously described (63–65). Sindbis and Semliki Forest viruses were propagated as previously described (66). Propagation of rVSV-LASV GP has been previously described (24).
Cloning and rescue of VSVs bearing hantavirus glycoproteins.
DNA sequences for the open reading frames (ORFs) of hantavirus glycoproteins, namely, Andes virus (ANDV) GP from the Chilean field strain R123 (GenBank accession number NP_604472.1), Sin Nombre virus (SNV) GP from a human field isolate (GenBank accession number NP_941974.1), and Hantaan virus (HTNV) GP from a cell culture-adapted strain, 76-118 (GenBank accession number NP_941978.1), were codon optimized for expression in human cells and synthesized by GenScript or Epoch Life Sciences Inc. The Ebola virus GP sequences in the vector encoding the rVSV-GPΔmuc (67) were replaced with these sequences by using standard molecular biology techniques. rVSV-ANDV GP, rVSV-SNV GP, and rVSV-HTNV GP viruses were generated by using a plasmid-based rescue system in 293T cells as described previously (68). Rescued viruses were amplified on Vero cells, and their identities were verified by sequencing of the hantaviral glycoprotein-encoding genes.
To generate fluorescent-protein-tagged virions, we genetically fused the fluorescent protein mNeonGreen (mNG) (69) to the VSV phosphoprotein (P) in a plasmid bearing VSV cDNA that lacks the G gene. DNA encoding mNG (GenBank accession number KC295282) was synthesized (Epoch Life Science) and fused to the N terminus of the VSV P ORF to generate VSV(mNG-P). This virus was rescued from cDNA and amplified on 293T cells by using published protocols (68, 70). Viral pseudotypes bearing ANDV GP were generated by infecting 293T cells expressing ANDV GP with VSV(mNG-P), as described previously (68).
Haploid genetic screen.
One hundred million mutagenized HAP1 cells were seeded and challenged with rVSV-ANDV GP (2 IU per cell) on the next day. Subsequently, surviving cells were harvested 8 days postinfection for extraction of genomic DNA. Genomic DNA was isolated using a DNA minikit (Qiagen), digested with NlaIII and MseI restriction enzymes, and subjected to linear amplification PCR, linker ligation, and exponential PCR as described previously (24). Samples were subjected to deep sequencing on a HiSeq200 sequencer (Illumina). Sequencing data were analyzed as described previously (24), except that filtering of close reads was omitted. To score enrichment of inactivating insertions (in the sense orientation in introns or inserted into exonic sequences) in the selected rVSV-ANDV GP-challenged population (86,554 unique insertion sites mapping to genes), data were compared to a similarly sized control data set of unselected HAP1 cells (151,357 unique insertions affecting genes). Besides enrichment of disruptive gene trap insertions over the control population, insertion sites in the selected and unselected cell pools were analyzed for orientational bias relative to the transcriptional direction of the affected genes as described previously (24) and plotted onto RefSeq gene structures.
CRISPR/Cas9-mediated knockout of S1P in human U2OS cells.
We knocked out the S1P gene in the human osteosarcoma U2OS cell line by clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9-mediated genome editing as described earlier (31). Human S1P gene (GenBank accession number NM_003791.3) contains 23 exons and is translated into a 1,052-amino-acid-long protein. D218 (aspartate), H249 (histidine), and S414 (serine) together form the active site of the serine protease S1P. A CRISPR guide RNA (gRNA) to target 1,716 to 1,738 nucleotides (target sequence 5′ GGTGCTGGCGTGCGGGGTTCTGG 3′) located in exon 10 of the S1P mRNA was cloned in the gRNA cloning vector (Addgene plasmid 41824) and used to generate the knockout cell line. In a 6-well plate, U2OS cells (2 × 105 cells per well) were cotransfected with plasmid encoding human-codon-optimized endonuclease Cas9 (hCas9; Addgene plasmid 41815, 3.0 µg/well), the gRNA cloning vector (Addgene plasmid 41824, 0.5 µg/well) expressing the above-described S1P-specific gRNA, a red fluorescent protein (RFP) expression plasmid (to monitor transfection efficiency; 0.1 µg/well), and pMX-IRES-Bla (confers blasticidin resistance to transfected cells; 0.4 µg/well) using Lipofectamine LTX (Life Technologies) as per the manufacturer’s instructions. At 24 h post-transfection, transfected cells were treated with 50 µg/ml of blasticidin for 24 to 36 h to kill the untransfected cells, and survivors were allowed to recover.
Genomic DNA was isolated from the surviving cells by using AquaPreserve and ProSink (MultiTarget Pharmaceuticals) according to the manufacturer’s directions. Genomic DNA flanking the gRNA target site was amplified by PCR using the 2× Phusion high-fidelity (HF) PCR master mix with HF buffer (Thermo Scientific) and cycling conditions of 98°C for 1 min, followed by 35 cycles of 98°C for 15 s, 65°C for 15 s, and 72°C for 30 s, and a final extension at 72°C for 3 min with the following primers: 5′ AATCACAGTTCTAGCACATAAATGG 3′ (forward) and 5′ CAGACTAAAACCAGTACCTTCCT 3′ (reverse). Amplified PCR products were tested for indels (insertion-deletions) at the target site by using a Surveyor mutation detection kit for standard gel electrophoresis (Transgenomic Inc.) as per the manufacturer’s instructions. Once indels were confirmed, genomic DNA from multiple single-cell clones was isolated and the gRNA-targeted region was amplified. After cloning of the amplified PCR product into a TOPO-TA vector followed by Sanger sequencing of multiple TOPO-TA clones, we found that both the alleles were mutated. Knockout of the S1P gene was confirmed by reverse transcription (RT)-PCR using primers S1P-F (5′ GATGTGCTCTGGCAGATGGG 3′) and S1P-R (5′ TTTCACGCCAGAACCCCGC 3′), specific for the deleted sequence in the S1P-#1 knockout cell clone.
Reconstitution of S1P expression in S1P knockout U2OS cells.
The full-length ORF of the S1P gene was amplified by PCR from the cDNA of the U2OS cells generated by using the SuperScript III first-strand synthesis system for RT-PCR (Life Technologies). The 2× Phusion high-fidelity PCR master mix with HF buffer (Thermo Scientific) was used to amplify the S1P ORF using the primers 5′ CAGTGTGGTGGTACGTAGGAATTCGTCGACGCCACCATGAAGCTTGTCAACATCTGGC 3′ (forward) and 5′ GAGTTTCTGCTCGAGCGGCCGCGTACGCGTCACCGAAGGGGTCTTTGGC 3′ (reverse). Cycling conditions were 98°C for 1 min, followed by 35 cycles of 98°C for 15 s, 65°C for 15 s, and 72°C for 2 min, and a final extension at 72°C for 5 min. The amplified product was cloned into a modified pBabe-Puro vector to express a C-terminal cMYC-DDK tagged S1P. Stable cells expressing S1P in the S1P-#1 background were generated as described earlier (46). Expression of S1P in the reconstituted cells was confirmed by immunostaining with an anti-Flag M2 antibody (Sigma).
Authentic hantavirus infections.
Wild-type, S1P knockout (S1P-#1) and S1P-reconstituted (S1P knockout cells stably expressing Flag-tagged S1P) U2OS cells were seeded in 96-well plates and incubated overnight at 37°C. Cells were inoculated with ANDV (1 IU per cell) for 2 h at 37°C. Virus inoculum was removed, and cells were washed with phosphate-buffered saline (PBS). Fresh culture medium was added, and cells were incubated at 37°C in 5% CO2. At 72 h postinfection (hpi), cells were washed with PBS and fixed with 10% formalin. Cells were permeabilized with 0.2% Triton X-100 for 10 min at room temperature and blocked with 1% bovine serum albumin (BSA) at 37°C for 2 h. Cells were incubated sequentially with ANDV nucleocapsid-specific rabbit polyclonal antibody NR-9673 (2.5 µg/ml for 2 h at room temperature; BEI Resources) and goat anti-rabbit IgG-Alexa 488 (1 mg/ml for 1 h at room temperature; Life Technologies). Cell nuclei were stained with Hoechst 33342 and then washed with PBS and stored at 4°C. Images were acquired at 20 fields/well with a 20× objective lens on an Operetta high-content imaging device (PerkinElmer). Images were analyzed with a customized scheme built from image analysis functions present in Harmony software, and the percentage of infected cells was determined using the analysis functions.
HUVECs were seeded in 96-well plates and incubated overnight at 37°C in 5% CO2. Cells were treated with 5 or 10 µM of the S1P inhibitor (PF-429242) or with the 1% DMSO vehicle for 24 h. Culture medium was removed, and fresh compound was added prior to inoculation with HTNV or SNV (1 IU per cell). Cells were incubated at 37°C for 24 h and immunostained for HTNV (HTNV nucleocapsid-specific rabbit polyclonal antibody NR-12152 [BEI Resources]) or SNV (SNV nucleocapsid-specific rabbit polyclonal antibody NR-9674 [BEI Resources]) antigen expression, and images were analyzed as described above.
Cytotoxicity assay.
Cytotoxicity of the S1P inhibitor PF-429242 (Tocris Bioscience) on the HUVECs was estimated by using the WST-1 assay (Roche) as per the manufacturer’s instructions.
Preparation of cholesterol:cyclodextrin complexes.
Cholesterol (Sigma-Aldrich) and 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich) complexes were prepared as described previously (71). After complex formation, the solution was filtered through a 0.45-µm syringe filter to remove any residual crystals.
Clostridium perfringens θ toxin-based cholesterol staining assay.
The cholesterol-binding domain (D4) of the Clostridium perfringens θ toxin was fused in frame to a hexahistidine tag and to enhanced blue fluorescent protein 2 (eBFP2), essentially as described for a D4-GFP fusion protein (36). The resulting eBFP2-θD4 construct was expressed in Escherichia coli, and protein was partially purified by nickel chelation chromatography, as described previously (36). After elution from nickel beads, the protein was further purified on a PD-10 desalting column (GE Healthcare). For cholesterol staining, U2OS cells were treated with 25 µM PF-429242 or the 1% DMSO vehicle (Fisher Scientific) for 24 h, followed by mock treatment or cholesterol supplementation for 5 min with 1 mM cholesterol:CD complex. Cells were washed three times with Hanks’ balanced salt solution (HBSS) (Corning) and then incubated with eBFP2-θD4 in HBSS containing 0.1% BSA for 45 to 60 min in the dark to stain the plasma membrane and/or endosomal cholesterol. Cells were washed with HBSS before being imaged by fluorescence microscopy under a 63× oil immersion objective (numerical aperture, 1.4; Carl Zeiss).
S1P inhibitor treatment and virus infection.
U2OS cells were pretreated with various concentrations of PF-429242 or the 1% DMSO vehicle for 24 h. Cells were inoculated with rVSV-G, rVSV-ANDV GP, rVSV-LASV GP, rVSV-HTNV GP, or Sindbis virus and incubated at 37°C in 5% CO2. At 2 hpi, 20 mM NH4Cl was added to stop subsequent rounds of infection. At 12 to 16 hpi, cell nuclei were counterstained with Hoechst 33342 dye (Life Technologies) and cells were imaged by fluorescence microscopy on an Axio Observer inverted microscope. Enhanced GFP (eGFP) expression was used to score infection. For experiments testing the effect of cholesterol supplementation on infection, cells received a cholesterol or mock treatment, as described above, prior to infection.
For all the infection experiments, individual channel images were exported as TIFF files, and infected (eGFP-positive) cells and nuclei were enumerated using CellProfiler (72). Infected cells were normalized to per-cell nuclear counts. For each technical replicate, 5,000 to 10,000 cells were counted.
Virus attachment and infection assay.
For both of these assays, U2OS cells were treated with 25 µM PF-429242 or the 1% DMSO vehicle for 24 h before being supplemented with a 62.5 µM cholesterol:CD complex or nothing for 0 to 60 min at 37°C. rVSV-ANDV GP was labeled with 5 µg/ml of a lipophilic dye, functional-component spacer diacyl lipid (FSL)-fluorescein (Sigma-Aldrich), and bound to the cell surface by centrifugation (2,500 rpm for 60 min at 4°C) in serum-free medium. Cells were placed on ice and washed with cold PBS. For the attachment experiment, cells were then fixed with 4% paraformaldehyde (PFA), and surface-bound virus was analyzed by flow cytometry. For the infection experiments, cells were placed at 37°C for 1 h, followed by the addition of 20 mM NH4Cl to stop further rounds of infection. At 12 to 16 hpi, cells were scored for infection as described above.
Internalization assay.
U2OS cells were pretreated with the 1% DMSO vehicle or 25 µM S1P inhibitor (PF-429242) for 24 h. VSV(mNG-P)-ANDV GP and VSV(mNG-P)-G particles containing mNeonGreen-tagged phosphoproteins were bound to the cell surface by centrifugation as described above. Cells were washed with cold PBS while on ice and incubated at 37°C for 0 to 60 min in the presence of the drug and 20 mM NH4Cl. Cells were then placed back on ice and treated with 0.5% trypsin (Life Technologies) for 10 min (ANDV GP) or 40 min (VSV G) to remove surface-bound virus. Cells were washed with cold PBS again and fixed with 4% PFA, and the fluorescence from internalized virus was measured by flow cytometry.
Fusion infection assay.
U2OS cells were pretreated with the 1% DMSO vehicle or 25 µM S1P inhibitor, followed by supplementation with cholesterol (1 mM for 5 min) or a mock treatment. rVSV-ANDV GP or Sindbis virus particles encoding eGFP were bound to the cell surface as described above. Cells were washed with cold PBS while on ice. The PBS was replaced with DMEM-F12 medium supplemented with 10 mM HEPES at pH 5.5 or 7.0. Cells were incubated at 37°C for 1 min and then returned to ice. Cells were incubated overnight at 37°C in 5% CO2 in medium containing drug and 20 mM NH4Cl. Cells were scored for infection as described above at 12 to 16 hpi.
Virus cell-lipid mixing assay.
The virus cell-lipid mixing assay was performed as for the fusion infection assay except that virus was labeled with a lipophilic dye, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine 4-chlorobenzenesulfonate (DiD) (15 µM), by mixing and incubating the dye with virus at room temperature for 10 min. After binding of the virus (2.6 IU per cell) at 4°C and incubation of virus-bound cells to pH 5.5 or 7.0 at 37°C, cells were washed with cold PBS and fixed with 4% paraformaldehyde on ice prior to being imaged for the DiD dequenching signal by fluorescence microscopy under a 10× air objective on an Axio Observer inverted microscope. Nuclei were counterstained with Hoechst 33342. Individual channel images were exported as TIFF files, and total DiD intensity and nuclear counts (5,000 to 10,000 cells per image) were determined using CellProfiler. DiD intensity values per cell (total DiD intensity divided by the nuclear count) are reported in Fig. 7E (4 to 5 images per condition).
Liposome preparation.
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), sphingomyelin (brain, porcine), and cholesterol (ovine wool, >98%) were purchased from Avanti Polar Lipids, Inc. The DOPE, DOPC, sphingomyelin, and cholesterol were dissolved in chloroform and methanol (2:1) and mixed in ratios of 1:1:1:0, 1:1:1:0.55, 1:1:1:1.5, and 1:1:1:3, respectively. Liposomes were generated by the freeze-thawing and extrusion method published in the work of Chatterjee et al. (73) using filters with a 0.4-µm pore size. The lipid films were hydrated in morpholineethanesulfonic acid (MES)-saline buffer (0.1 M MES and 0.9% NaCl) (pH 8) and stored under nitrogen gas at 4°C.
Virus liposome-lipid mixing assay.
rVSVs (30 µg of virus, based on total protein) and Semliki Forest virus (7.5 µg of virus, based on total protein) were labeled with 15 µM and 7.5 µM DiD, respectively. Lipid mixing with liposomes was measured by DiD dequenching and recorded at excitation and emission wavelengths of 620 nm and 665 nm, respectively, on an Aminco Bowman series 2 luminescence spectrometer. Each assay mixture contained 2 ml of HNE buffer (5 mM HEPES, 150 mM NaCl, and 0.1 mM EDTA, pH 7.3) along with 0.2 mM liposomes and pretitrated amounts of DiD-labeled virus. The experiments were performed under constant stirring in a quartz cuvette at 37°C. Pretitrated amounts of 0.25 M MES (pH 4.8) were injected into the samples to achieve pH 7.3 or 5.5. After fluorescence was recorded for several minutes, Triton X-100 was added to a final concentration of 0.1%. For normalization, the minimum fluorescence value was taken as zero, and the maximum fluorescence obtained by adding Triton X-100 was set to 100.
VSV M protein release assay.
This assay was performed using rVSV-G or rVSV-ANDV GP on WT or S1P-null U2OS cells as described previously (22, 46). Briefly, U2OS cells were pretreated with 20 µg/ml of cycloheximide (Acros Organics) for 30 min and infected with rVSV-ANDV GP or rVSV-G (200 IU per cell) in the presence of cycloheximide for 3 h at 37°C. Cells were washed with PBS and fixed and immunostained for VSV M antigen. Cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) and visualized by fluorescence microscopy under a 20× air objective on an Axio Observer inverted microscope (Zeiss). Individual channel images were exported as TIFF files, and puncta containing VSV M protein and cell nuclei were enumerated using CellProfiler. For each sample, approximately 1,000 cells from 11 to 12 microscopic fields were counted.
SUPPLEMENTAL MATERIAL
The S1P inhibitor exhibits marginal cytotoxicity in human umbilical vein endothelial cells (HUVEC). HUVEC were treated with the 1% DMSO vehicle (as a control) or S1P inhibitor (3, 6, or 12 µM) for 24 h, and metabolic activity was measured by using a colorimetric assay (WST-1 assay) at 405 nm. Download
Knockout of the S1P gene delays, but does not block, viral internalization. WT or S1P-null U2OS cells were exposed to VSV(mNG-P)-ANDV GP (left) or VSV(mNG-P)-G (right) particles at 4°C. Following internalization of virus at 37°C for the indicated times, remaining surface-bound virus particles were removed by a brief trypsin treatment. Cells were harvested and fixed with 4% PFA, and internalized virus was measured by flow cytometry (n = 2; two independent experiments). Download
ANDV GP-mediated fusion infection is most efficient at pH 5.5. U2OS cells were exposed to rVSV-ANDV GP viruses (2.2 IU per cell). After binding of virus to the cells at 4°C, cells were exposed to the indicated pH medium at 37°C for 1 min and then returned to medium containing 20 mM NH4Cl. Cells were incubated at 37°C overnight and scored for infection the next day (n = 2). Download
Alphavirus membrane fusion and infection are less affected by S1P inhibitor-mediated cholesterol depletion than those of rVSV-ANDV GP. (A) Fusion infection with the alphavirus Sindbis virus (0.5 IU per cell) was carried out as described for Fig. 7C (n = 4; two independent experiments). (B) U2OS cells pretreated with 25 µM S1P inhibitor PF-429242 or the 1% DMSO vehicle for 24 h were exposed to rVSV-ANDV GP or Sindbis virus (0.5 IU per cell). After 1 h of incubation, NH4Cl (20 mM, final concentration) was added to the medium to prevent subsequent rounds of infection. Infected cells were enumerated as described for Fig. 2 (n = 4; two independent experiments). Download
Detection of DiD dequenching of labeled rVSV particles. rVSV-G particles were labeled with 15 µM DiD and serially diluted in the presence or absence of 0.1% Triton X-100. DiD fluorescence was recorded using a fluorescence plate reader at excitation and emission wavelengths of 644 nm and 670 nm, respectively. Download
ANDV GP-mediated cytoplasmic release of VSV M protein is greatly reduced in S1P-null cells. WT and S1P-null U2OS cells pretreated with cycloheximide were exposed to rVSV-ANDV GP or rVSV-G particles (200 IU per cell) for 3 h at 37°C. Cells were fixed and immunostained for VSV M protein. (A) Representative images (from one out of multiple independent experiments) showing VSV M protein (red) and cell nuclei (blue). (B) Puncta containing VSV M protein were enumerated and are represented here as the average number of puncta per cell for each microscopic field (11 to 12 fields). Download
ACKNOWLEDGMENTS
We thank Tyler Krause for technical support. We are grateful to V. R. Prasad and T. Y. Chang for useful discussions.
This work was supported by NIH grants R01 AI088027 (to K.C.) and R01 GM057454 (to M.K.), a JSTO-CBD Defense Threat Reduction Agency grant from project CB3947 (to J.M.D.), and a European Research Council (ERC) starting grant (ERC-2012-StG 309634) (to T.R.B.). K.C. was additionally supported by a fellowship from the Irma T. Hirschl/Monique Weill-Caulier Trust and by a Harold and Muriel Block Faculty Scholarship at the Albert Einstein College of Medicine. L.M.K. was additionally supported by the NIH Training Program in Cellular and Molecular Biology and Genetics (grant T32 GM007491).
Opinions, conclusions, interpretations, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Department of the Army, the U.S. Department of Defense, or the U.S. Department of Health and Human Services.
Footnotes
Citation Kleinfelter LM, Jangra RK, Jae LT, Herbert AS, Mittler E, Stiles KM, Wirchnianski AS, Kielian M, Brummelkamp TR, Dye JM, Chandran K. 2015. Haploid genetic screen reveals a profound and direct dependence on cholesterol for hantavirus membrane fusion. mBio 6(4):e00801-15. doi:10.1128/mBio.00801-15.
REFERENCES
- 1.Peters CJ, Simpson GL, Levy H. 1999. Spectrum of hantavirus infection: hemorrhagic fever with renal syndrome and hantavirus pulmonary syndrome. Annu Rev Med 50:531–545. doi: 10.1146/annurev.med.50.1.531. [DOI] [PubMed] [Google Scholar]
- 2.Schmaljohn C, Hjelle B. 1997. Hantaviruses: a global disease problem. Emerg Infect Dis 3:95–104. doi: 10.3201/eid0302.970202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaheri A, Strandin T, Hepojoki J, Sironen T, Henttonen H, Mäkelä S, Mustonen J. 2013. Uncovering the mysteries of hantavirus infections. Nat Rev Microbiol 11:539–550. doi: 10.1038/nrmicro3066. [DOI] [PubMed] [Google Scholar]
- 4.Lee HW. 1996. Epidemiology and pathogenesis of hemorrhagic fever with renal syndrome, p 253–267. In Elliott RM (ed), The Bunyaviridae. Springer US, Boston, MA. [Google Scholar]
- 5.MacNeil A, Nichol ST, Spiropoulou CF. 2011. Hantavirus pulmonary syndrome. Virus Res 162:138–147. doi: 10.1016/j.virusres.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 6.Núñez JJ, Fritz CL, Knust B, Buttke D, Enge B, Novak MG, Kramer V, Osadebe L, Messenger S, Albariño CG, Ströher U, Niemela M, Amman BR, Wong D, Manning CR, Nichol ST, Rollin PE, Xia D, Watt JP, Vugia DJ, Yosemite Hantavirus Outbreak Investigation Team . 2014. Hantavirus infections among overnight visitors to Yosemite. National Park, California, USA, 2012. Emerg Infect Dis 20:386–393. doi: 10.3201/eid2003.131581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott RM, Schmaljohn CS. 2014. Bunyaviridae, p 1244–1282. In Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 8.Schmaljohn CS, Schmaljohn AL, Dalrymple JM. 1987. Hantaan virus M RNA: coding strategy, nucleotide sequence, and gene order. Virology 157:31–39. doi: 10.1016/0042-6822(87)90310-2. [DOI] [PubMed] [Google Scholar]
- 9.Kamrud KI, Schmaljohn CS. 1994. Expression strategy of the M genome segment of Hantaan virus. Virus Res 31:109–121. doi: 10.1016/0168-1702(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 10.Löber C, Anheier B, Lindow S, Klenk HD, Feldmann H. 2001. The Hantaan virus glycoprotein precursor is cleaved at the conserved pentapeptide WAASA. Virology 289:224–229. doi: 10.1006/viro.2001.1171. [DOI] [PubMed] [Google Scholar]
- 11.Ruusala A, Persson R, Schmaljohn CS, Pettersson RF. 1992. Coexpression of the membrane glycoproteins G1 and G2 of Hantaan virus is required for targeting to the Golgi complex. Virology 186:53–64. doi: 10.1016/0042-6822(92)90060-3. [DOI] [PubMed] [Google Scholar]
- 12.Deyde VM, Rizvanov AA, Chase J, Otteson EW, St Jeor SC. 2005. Interactions and trafficking of Andes and sin Nombre hantavirus glycoproteins G1 and G2. Virology 331:307–315. doi: 10.1016/j.virol.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Antic D, Wright KE, Kang CY. 1992. Maturation of Hantaan virus glycoproteins G1 and G2. Virology 189:324–328. doi: 10.1016/0042-6822(92)90709-X. [DOI] [PubMed] [Google Scholar]
- 14.Cifuentes-Muñoz N, Salazar-Quiroz N, Tischler ND. 2014. Hantavirus Gn and Gc envelope glycoproteins: key structural units for virus cell entry and virus assembly. Viruses 6:1801–1822. doi: 10.3390/v6041801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. 1998. β3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc Natl Acad Sci U S A 95:7074–7079. doi: 10.1073/pnas.95.12.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER. 1999. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by β3 integrins. J Virol 73:3951–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackow ER, Gavrilovskaya IN. 2001. Cellular receptors and hantavirus pathogenesis. Curr Top Microbiol Immunol 256:91–115. doi: 10.1007/978-3-642-56753-7_6. [DOI] [PubMed] [Google Scholar]
- 18.Krautkrämer E, Zeier M. 2008. Hantavirus causing hemorrhagic fever with renal syndrome enters from the apical surface and requires decay-accelerating factor (DAF/CD55). J Virol 82:4257–4264. doi: 10.1128/JVI.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi Y, Kwon YC, Kim SI, Park JM, Lee KH, Ahn BY. 2008. A hantavirus causing hemorrhagic fever with renal syndrome requires gC1qR/p32 for efficient cell binding and infection. Virology 381:178–183. doi: 10.1016/j.virol.2008.08.035. [DOI] [PubMed] [Google Scholar]
- 20.Raftery MJ, Lalwani P, Krautkrmer E, Peters T, Scharffetter-Kochanek K, Krüger R, Hofmann J, Seeger K, Krüger DH, Schönrich G. 2014. β2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J Exp Med 211:1485–1497. doi: 10.1084/jem.20131092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petersen J, Drake MJ, Bruce EA, Riblett AM, Didigu CA, Wilen CB, Malani N, Male F, Lee F-H, Bushman FD, Cherry S, Doms RW, Bates P, Briley K. 2014. The major cellular sterol regulatory pathway is required for Andes virus infection. PLoS Pathog 10:e1003911. doi: 10.1371/journal.ppat.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, Brummelkamp TR. 2009. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 24.Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P, Wessels MW, Lefeber DJ, Whelan SP, van Bokhoven H, Brummelkamp TR. 2013. Deciphering the glycosylome of dystroglycanopathies using haploid screens for Lassa virus entry. Science 340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, Whelan SP, Dye JM, Brummelkamp TR. 2014. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakai J, Nohturfft A, Goldstein JL, Brown MS. 1998. Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. J Biol Chem 273:5785–5793. doi: 10.1074/jbc.273.10.5785. [DOI] [PubMed] [Google Scholar]
- 27.Brown MS, Goldstein JL. 1999. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A 96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown MS, Goldstein JL. 1997. The SREBP pathway: regulation review of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89:331–340. doi: 10.1016/S0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 29.Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL. 1996. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85:1037–1046. doi: 10.1016/S0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- 30.Hua X, Sakai J, Brown MS, Goldstein JL. 1996. Regulated cleavage of sterol regulatory element binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J Biol Chem 271:10379–10384. doi: 10.1074/jbc.271.17.10379. [DOI] [PubMed] [Google Scholar]
- 31.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hawkins JL, Robbins MD, Warren LC, Xia D, Petras SF, Valentine JJ, Varghese AH, Wang I-K, Subashi TA, Shelly LD, Hay BA, Landschulz KT, Geoghegan KF, Harwood HJ. 2008. Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J Pharmacol Exp Ther 326:801–808. doi: 10.1124/jpet.108.139626. [DOI] [PubMed] [Google Scholar]
- 33.Hay BA, Abrams B, Zumbrunn AY, Valentine JJ, Warren LC, Petras SF, Shelly LD, Xia A, Varghese AH, Hawkins JL, Van Camp JA, Robbins MD, Landschulz K, Harwood HJ. 2007. Aminopyrrolidineamide inhibitors of site-1 protease. Bioorg Med Chem Lett 17:4411–4414. doi: 10.1016/j.bmcl.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 34.Sieczkarski SB, Whittaker GR. 2003. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 35.Pasqual G, Rojek JM, Masin M, Chatton J-Y, Kunz S. 2011. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog 7:e1002232. doi: 10.1371/journal.ppat.1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimada Y, Maruya M, Iwashita S, Ohno-Iwashita Y. 2002. The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur J Biochem 269:6195–6203. doi: 10.1046/j.1432-1033.2002.03338.x. [DOI] [PubMed] [Google Scholar]
- 37.Rawson RB, DeBose-Boyd R, Goldstein JL, Brown MS. 1999. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J Biol Chem 274:28549–28556. doi: 10.1074/jbc.274.40.28549. [DOI] [PubMed] [Google Scholar]
- 38.Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL, Brown MS. 1998. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell 2:505–514. doi: 10.1016/S1097-2765(00)80150-1. [DOI] [PubMed] [Google Scholar]
- 39.Jin M, Park J, Lee S, Park B, Shin J, Song K-J, Ahn T-I, Hwang S-Y, Ahn B-Y, Ahn K. 2002. Hantaan virus enters cells by clathrin-dependent receptor-mediated endocytosis. Virology 294:60–69. doi: 10.1006/viro.2001.1303. [DOI] [PubMed] [Google Scholar]
- 40.Ramanathan HN, Jonsson CB. 2008. New and Old World hantaviruses differentially utilize host cytoskeletal components during their life cycles. Virology 374:138–150. doi: 10.1016/j.virol.2007.12.030. [DOI] [PubMed] [Google Scholar]
- 41.Lozach P-Y, Mancini R, Bitto D, Meier R, Oestereich L, Overby AK, Pettersson RF, Helenius A. 2010. Entry of bunyaviruses into mammalian cells. Cell Host Microbe 7:488–499. doi: 10.1016/j.chom.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu YE, Cassese T, Kielian M. 1999. The cholesterol requirement for Sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J Virol 73:4272–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825. doi: 10.3390/v2040796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chernomordik LV, Kozlov MM. 2008. Mechanics of membrane fusion. Nat Struct Mol Biol 15:675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kielian M. 2014. Mechanisms of virus membrane fusion proteins. Annu Rev Virol 1:171–189. doi: 10.1146/annurev-virology-031413-085521. [DOI] [PubMed] [Google Scholar]
- 46.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. 2012. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phalen T, Kielian M. 1991. Cholesterol is required for infection by Semliki forest virus. J Cell Biol 112:615–623. doi: 10.1083/jcb.112.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, Ploegh HL, Brummelkamp TR. 2011. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol 29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cifuentes-Muñoz N, Darlix J-L, Tischler ND. 2010. Development of a lentiviral vector system to study the role of the Andes virus glycoproteins. Virus Res 153:29–35. doi: 10.1016/j.virusres.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Bergeron E, Vincent MJ, Nichol ST. 2007. Crimean-Congo hemorrhagic fever virus glycoprotein processing by the endoprotease SKI-1/S1P is critical for virus infectivity. J Virol 81:13271–13276. doi: 10.1128/JVI.01647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lenz O, ter Meulen J, Klenk HD, Seidah NG, Garten W. 2001. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc Natl Acad Sci U S A 98:12701–12705. doi: 10.1073/pnas.221447598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beyer WR, Pöpplau D, Garten W, Laer von D, Lenz O. 2003. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J Virol 77:2866–2872. doi: 10.1128/JVI.77.5.2866-2872.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugii S, Lin S, Ohgami N, Ohashi M, Chang CC, Chang T-Y. 2006. Roles of endogenously synthesized sterols in the endocytic pathway. J Biol Chem 281:23191–23206. doi: 10.1074/jbc.M603215200. [DOI] [PubMed] [Google Scholar]
- 54.Danthi P, Chow M. 2004. Cholesterol removal by methyl-beta-cyclodextrin inhibits poliovirus entry. J Virol 78:33–41. doi: 10.1128/JVI.78.1.33-41.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hullin-Matsuda F, Taguchi T, Greimel P, Kobayashi T. 2014. Lipid compartmentalization in the endosome system. Semin Cell Dev Biol 31:48–56. doi: 10.1016/j.semcdb.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 56.Van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi T, Beuchat M-H, Chevallier J, Makino A, Mayran N, Escola J-M, Lebrand C, Cosson P, Kobayashi T, Gruenberg J. 2002. Separation and characterization of late endosomal membrane domains. J Biol Chem 277:32157–32164. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 58.Möbius W, van Donselaar E, Ohno-Iwashita Y, Shimada Y, Heijnen HF, Slot JW, Geuze HJ. 2003. Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 4:222–231. doi: 10.1034/j.1600-0854.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 59.Sobo K, Le Blanc I, Luyet P-P, Fivaz M, Ferguson C, Parton RG, Gruenberg J, van der Goot FG. 2007. Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS One 2:e851. doi: 10.1371/journal.pone.0000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tischler ND, Gonzalez A, Perez-Acle T, Rosemblatt M, Valenzuela PD. 2005. Hantavirus Gc glycoprotein: evidence for a class II fusion protein. J Gen Virol 86:2937–2947. doi: 10.1099/vir.0.81083-0. [DOI] [PubMed] [Google Scholar]
- 61.Needham D, Nunn RS. 1990. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys J 58:997–1009. doi: 10.1016/S0006-3495(90)82444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Papanikolaou A, Papafotika A, Murphy C, Papamarcaki T, Tsolas O, Drab M, Kurzchalia TV, Kasper M, Christoforidis S. 2005. Cholesterol-dependent lipid assemblies regulate the activity of the ecto-nucleotidase CD39. J Biol Chem 280:26406–26414. doi: 10.1074/jbc.M413927200. [DOI] [PubMed] [Google Scholar]
- 63.Lee HW, Lee PW, Johnson KM. 1978. Isolation of the etiologic agent of Korean hemorrhagic fever. J Infect Dis 137:298–308. doi: 10.1093/infdis/137.3.298. [DOI] [PubMed] [Google Scholar]
- 64.Schmaljohn AL, Li D, Negley DL, Bressler DS, Turell MJ, Korch GW, Ascher MS, Schmaljohn CS. 1995. Isolation and initial characterization of a newfound hantavirus from California. Virology 206:963–972. doi: 10.1006/viro.1995.1019. [DOI] [PubMed] [Google Scholar]
- 65.Hooper JW, Larsen T, Custer DM, Schmaljohn CS. 2001. A lethal disease model for hantavirus pulmonary syndrome. Virology 289:6–14. doi: 10.1006/viro.2001.1133. [DOI] [PubMed] [Google Scholar]
- 66.Glomb-Reinmund S, Kielian M. 1998. The role of low pH and disulfide shuffling in the entry and fusion of Semliki Forest virus and Sindbis virus. Virology 248:372–381. doi: 10.1006/viro.1998.9275. [DOI] [PubMed] [Google Scholar]
- 67.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. 2010. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J Virol 84:163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A 92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J. 2013. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klein U, Gimpl G, Fahrenholz F. 1995. Alteration of the myometrial plasma membrane cholesterol content with β-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- 72.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chatterjee PK, Vashishtha M, Kielian M. 2000. Biochemical consequences of a mutation that controls the cholesterol dependence of Semliki Forest virus fusion. J Virol 74:1623–1631. doi: 10.1128/JVI.74.4.1623-1631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The S1P inhibitor exhibits marginal cytotoxicity in human umbilical vein endothelial cells (HUVEC). HUVEC were treated with the 1% DMSO vehicle (as a control) or S1P inhibitor (3, 6, or 12 µM) for 24 h, and metabolic activity was measured by using a colorimetric assay (WST-1 assay) at 405 nm. Download
Knockout of the S1P gene delays, but does not block, viral internalization. WT or S1P-null U2OS cells were exposed to VSV(mNG-P)-ANDV GP (left) or VSV(mNG-P)-G (right) particles at 4°C. Following internalization of virus at 37°C for the indicated times, remaining surface-bound virus particles were removed by a brief trypsin treatment. Cells were harvested and fixed with 4% PFA, and internalized virus was measured by flow cytometry (n = 2; two independent experiments). Download
ANDV GP-mediated fusion infection is most efficient at pH 5.5. U2OS cells were exposed to rVSV-ANDV GP viruses (2.2 IU per cell). After binding of virus to the cells at 4°C, cells were exposed to the indicated pH medium at 37°C for 1 min and then returned to medium containing 20 mM NH4Cl. Cells were incubated at 37°C overnight and scored for infection the next day (n = 2). Download
Alphavirus membrane fusion and infection are less affected by S1P inhibitor-mediated cholesterol depletion than those of rVSV-ANDV GP. (A) Fusion infection with the alphavirus Sindbis virus (0.5 IU per cell) was carried out as described for Fig. 7C (n = 4; two independent experiments). (B) U2OS cells pretreated with 25 µM S1P inhibitor PF-429242 or the 1% DMSO vehicle for 24 h were exposed to rVSV-ANDV GP or Sindbis virus (0.5 IU per cell). After 1 h of incubation, NH4Cl (20 mM, final concentration) was added to the medium to prevent subsequent rounds of infection. Infected cells were enumerated as described for Fig. 2 (n = 4; two independent experiments). Download
Detection of DiD dequenching of labeled rVSV particles. rVSV-G particles were labeled with 15 µM DiD and serially diluted in the presence or absence of 0.1% Triton X-100. DiD fluorescence was recorded using a fluorescence plate reader at excitation and emission wavelengths of 644 nm and 670 nm, respectively. Download
ANDV GP-mediated cytoplasmic release of VSV M protein is greatly reduced in S1P-null cells. WT and S1P-null U2OS cells pretreated with cycloheximide were exposed to rVSV-ANDV GP or rVSV-G particles (200 IU per cell) for 3 h at 37°C. Cells were fixed and immunostained for VSV M protein. (A) Representative images (from one out of multiple independent experiments) showing VSV M protein (red) and cell nuclei (blue). (B) Puncta containing VSV M protein were enumerated and are represented here as the average number of puncta per cell for each microscopic field (11 to 12 fields). Download