

ABSTRACT
The motile-to-sessile transition is an important lifestyle switch in diverse bacteria and is often regulated by the intracellular second messenger cyclic diguanylate monophosphate (c-di-GMP). In general, high c-di-GMP concentrations promote attachment to surfaces, whereas cells with low levels of signal remain motile. In the plant pathogen Agrobacterium tumefaciens, c-di-GMP controls attachment and biofilm formation via regulation of a unipolar polysaccharide (UPP) adhesin. The levels of c-di-GMP in A. tumefaciens are controlled in part by the dual-function diguanylate cyclase-phosphodiesterase (DGC-PDE) protein DcpA. In this study, we report that DcpA possesses both c-di-GMP synthesizing and degrading activities in heterologous and native genetic backgrounds, a binary capability that is unusual among GGDEF-EAL domain-containing proteins. DcpA activity is modulated by a pteridine reductase called PruA, with DcpA acting as a PDE in the presence of PruA and a DGC in its absence. PruA enzymatic activity is required for the control of DcpA and through this control, attachment and biofilm formation. Intracellular pterin analysis demonstrates that PruA is responsible for the production of a novel pterin species. In addition, the control of DcpA activity also requires PruR, a protein encoded directly upstream of DcpA with a predicted molybdopterin-binding domain. PruR is hypothesized to be a potential signaling intermediate between PruA and DcpA through an as-yet-unidentified mechanism. This study provides the first prokaryotic example of a pterin-mediated signaling pathway and a new model for the regulation of dual-function DGC-PDE proteins.
IMPORTANCE
Pathogenic bacteria often attach to surfaces and form multicellular communities called biofilms. Biofilms are inherently resilient and can be difficult to treat, resisting common antimicrobials. Understanding how bacterial cells transition to the biofilm lifestyle is essential in developing new therapeutic strategies. We have characterized a novel signaling pathway that plays a dominant role in the regulation of biofilm formation in the model pathogen Agrobacterium tumefaciens. This control pathway involves small metabolites called pterins, well studied in eukaryotes, but this is the first example of pterin-dependent signaling in bacteria. The described pathway controls levels of an important intracellular second messenger (cyclic diguanylate monophosphate) that regulates key bacterial processes such as biofilm formation, motility, and virulence. Pterins control the balance of activity for an enzyme that both synthesizes and degrades the second messenger. These findings reveal a complex, multistep pathway that modulates this enzyme, possibly identifying new targets for antibacterial intervention.
INTRODUCTION
Bacterial cells often exist as members of multicellular communities known as biofilms, which are commonly formed by both commensal and pathogenic bacteria (1, 2). Bacteria within a biofilm are encased within a complex extracellular matrix (3) that often provides protection against toxic substances (4, 5) and desiccation (6) and facilitates nutrient exchange (7). Biofilms are difficult to control due to their intrinsic antibiotic tolerance (8). Therapeutic intervention targeting bacterial attachment and the subsequent steps that lead to biofilm formation could hold promise in treating a variety of bacterial infections. Understanding how these bacteria orchestrate the transition from a motile, free-living lifestyle to a sessile, multicellular biofilm is a major goal of recent research.
Cyclic diguanylate monophosphate (c-di-GMP) is a nucleotide second messenger (9, 10) that plays a critical role in the regulation of bacterial attachment and biofilm formation in diverse bacteria. Since its discovery over 25 years ago (11), c-di-GMP has been implicated in controlling processes intimately associated with biofilm formation such as polysaccharide biosynthesis (12, 13), production and transport of biofilm matrix components (14, 15), oxygen-dependent regulation (16), and motility (17). In addition, c-di-GMP can also control virulence (18, 19) and morphological development (20, 21). Intracellular c-di-GMP levels are perceived through several varieties of c-di-GMP-responsive receptors, enzyme allosteric sites, transcription factors, and riboswitches (19, 22–25). Elevated levels of c-di-GMP typically favor the sessile state, resulting in increased attachment and biofilm formation, whereas lower levels of c-di-GMP favor the motile, planktonic state and the downregulation of attachment (10).
The c-di-GMP concentration within a cell is controlled by the opposing activities of diguanylate cyclases (DGCs) and phosphodiesterases (PDEs), which possess synthetic and degradative activities, respectively (10, 26). DGCs are characterized by the GGDEF catalytic motif (21), and PDEs are characterized by either an EAL (27, 28) or HD-GYP catalytic motif (29). Proteins with both GGDEF and EAL domains are common, but in many examples, one of the domains is catalytically inactive (30, 31). There are few examples of enzymes that are known to have both DGC and PDE activities (32–35), and their in vivo regulation and activity are poorly understood (36–38).
Agrobacterium tumefaciens is a facultative plant pathogen that causes the neoplastic disease called crown gall via cross-kingdom horizontal gene transfer and integration of plasmid-derived tumorigenic DNA into the plant genome (39–41). A. tumefaciens forms biofilms on both biotic and abiotic surfaces (42, 43), with attachment to the plant surface as a required component of plant transformation. Attachment to a range of surfaces is dependent upon the unipolar polysaccharide (UPP) adhesin (44, 45) with additional influence from cellulose (46, 47). Regulation of A. tumefaciens attachment is controlled by multiple integrated regulatory pathways (48), several of which play a role in modulating intracellular c-di-GMP levels.
Shortly after its original discovery, the presence of c-di-GMP was detected in A. tumefaciens, and its role in the regulation of cellulose synthesis was described (13). This regulation was recently attributed, at least in part, to a DGC in A. tumefaciens (49, 50) that is homologous to the well-studied PleD GGDEF protein from Caulobacter crescentus. The levels of c-di-GMP have also been shown to directly regulate attachment, with elevated c-di-GMP concentrations driven by overexpression of the A. tumefaciens PleD leading to increased levels of UPP production, cellulose synthesis, and biofilm formation (50). Despite these observations, little is known about how c-di-GMP signaling is integrated into the overall regulatory network for controlling attachment and biofilm formation in A. tumefaciens.
In the search for regulatory candidates, a transposon mutagenesis screen was performed for mutants that mimicked the elevated UPP production phenotype of a PleD-overexpressing strain (50). Several classes of transposon mutations were described, including the visNR locus, encoding the master motility regulators in A. tumefaciens, now known to inhibit c-di-GMP synthesis through two DGC proteins that do not include PleD. Two other intriguing mutant classes included a putative pteridine reductase (Atu1130, designated here as PruA) and a dual GGDEF-EAL protein (Atu3495, now designated DcpA [diguanylate cyclase/phosphodiesterase A]). Transposon mutations in either dcpA or pruA led to increased staining with the polysaccharide-reactive dye Congo red (reporting on UPP and cellulose production) and elevated attachment and biofilm formation (50), suggesting that these genes are negative regulators of A. tumefaciens surface interactions. It was hypothesized that a loss of DcpA PDE function might lead to increased UPP and cellulose production through elevated levels of c-di-GMP.
In this study, the PDE activity of DcpA is shown to be necessary for the negative regulation of attachment. However, DcpA can also act as a DGC, implicating DcpA as a dual-function DGC-PDE protein. The primary state of DcpA activity in A. tumefaciens is regulated via a complex control pathway that involves the production of a low-molecular-weight metabolite known as a pterin, a class of redox-reactive enzymatic prosthetic groups, by a putative pteridine reductase (PruA). This control of DcpA influences UPP and cellulose production, attachment, and biofilm formation of A. tumefaciens. The pterin produced by PruA requires a putative pterin-binding protein PruR, encoded immediately upstream of DcpA, to influence DcpA activity, establishing a mechanism by which pterin-dependent signaling modulates the balance between motile to sessile growth modes for A. tumefaciens.
RESULTS
Genetic and phenotypic evidence for DcpA phosphodiesterase and diguanylate cyclase activity.
Our previous studies suggested that the Atu3495 gene product (now designated DcpA), which contains highly conserved GGDEF and EAL domains, acts as a phosphodiesterase (PDE) in wild-type A. tumefaciens based on several surface attachment-associated phenotypes of a dcpA in-frame deletion mutant (50). The DcpA coding sequence is 1,935 bp and is located on the A. tumefaciens C58 linear chromosome, 8 bp downstream of a predicted open reading frame coding for the conserved hypothetical protein Atu3496. Hence, these two genes are likely to form an operon (Fig. 1A). Atu3496 and its potential relationship to DcpA will be described below. Upstream of the Atu3496 gene and separated by 181 bp is the Atu3497 gene, which encodes a conserved hypothetical protein with no recognized domains.
FIG 1 .

Complementation of increased biofilm formation in the A. tumefaciens dcpA mutant requires an intact DcpA EAL catalytic motif. (A) Diagram of PruR-DcpA genetic locus. Atu3497 is a conserved hypothetical protein with no annotated domains. (B) Protein topology of DcpA. Protein domains predicted by the BLAST database (NCBI) are shown. Domains are drawn to scale. TM, transmembrane; DGC, diguanylate cyclase; PDE, phosphodiesterase. (C) A. tumefaciens quantitative biofilm formation on PVC coverslips after 48 h of static growth at 28°C. Adherent biomass was quantified by staining with crystal violet (CV). CV absorbance was quantified by absorbance at 600 nm (A600). In parallel, the optical density at 600 nm (OD600) of planktonic culture was determined. CV absorbance was normalized to culture growth by calculating the A600/OD600 ratio. IPTG (400 µM) was added to all strains. The wild-type (WT) strain, ΔdcpA mutant with no plasmid inserted (-), and ΔdcpA mutant strain with Plac-dcpA derivatives are shown. A wild-type copy of dcpA provided on a plasmid expressed from the lacZ promoter (Plac-dcpA; DGC+PDE+) was introduced into a ΔdcpA mutant. The Plac-dcpA derivatives include a DcpA variant containing catalytic site GGDEF→GGDAF mutation (DGC−) and a DcpA variant containing catalytic site EAL→AAL mutation (PDE−). Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 standard deviation (SD).
The N terminus of DcpA contains a predicted periplasmic region of approximately 140 amino acid (aa) residues flanked by two transmembrane domains (Fig. 1B). It is possible that this periplasmic segment plays a sensory function, but the region is not homologous to any known protein domains. A predicted DGC domain (residues 226 to 381) lies carboxy terminal to the periplasmic domain. This domain contains a GGDEF motif and other conserved residues needed for proper enzymatic activity of canonical DGC proteins, such as those involved in GTP and metal binding (see Fig. S1A in the supplemental material) (23, 51). Notably, DcpA lacks a conserved RXXD I-site motif, which is normally involved in negative allosteric feedback of c-di-GMP synthesis (23). The PDE domain of DcpA (residues 400 to 672) is composed of a canonical EAL catalytic motif and other essential residues necessary for metal coordination (Fig. S1B). In summary, DcpA has all the necessary residues consistent with enzymatically active DGC and PDE domains and is named dcpA (for diguanylate cyclase/phosphodiesterase A).
In order to delve further into the mechanistic basis for the aforementioned phenotypes and to genetically test DcpA’s role as a PDE, a targeted mutation (E431A) was constructed to change the EAL catalytic motif to AAL. In parallel and in combination with the EAL mutation, the GGDEF catalytic motif of DcpA was mutated to GGDAF (E308A). These mutations have been reported to enzymatically inactivate PDE domains and DGC domains, respectively (26, 27, 50, 52).
A wild-type copy of dcpA provided on a plasmid expressed from the lacZ promoter (Plac-dcpA; DGC+PDE+) was introduced into a ΔdcpA mutant, and when it was induced with isopropyl-β-d-thiogalactopyranoside (IPTG), it diminished biofilm formation of the ΔdcpA mutant (Fig. 1C, P value of <0.05 by paired t test) and Congo red staining, reflecting polysaccharide levels, to near wild-type (WT) levels (see Fig. S2A in the supplemental material). Decreased biofilm formation was responsive to increasing levels of IPTG (Fig. S2B). Mutating the GGDEF catalytic motif (E308A) had no effect on the ability of dcpA to complement the null mutation (Fig. 1C, P value of <0.05 by paired t test compared to the value for the WT). However, mutation of the EAL motif in the PDE domain (E431A) alone abolished the ability of dcpA to complement the mutant biofilm phenotype back to WT levels, and this plasmid in fact stimulated biofilm formation approximately 10-fold greater than that observed in the ΔdcpA mutant (Fig. 1C, P value of <0.05 by paired t test). This elevated level of biofilm formation is due to a functional GGDEF motif, as the plasmid-borne dcpA double mutant (E308A E431A) did not elevate biofilm levels above the ΔdcpA mutant (Fig. 1C). The phenotypes of the PDE disabled dcpA variant were not multicopy effects, as the mutation introduced into the native copy of dcpA resulted in the same phenotypes as the plasmid-borne allele (Fig. S2A and data not shown). Direct measurement of c-di-GMP in whole-cell extracts revealed that the ΔdcpA mutant tended to be slightly elevated, but these values were not statistically different from wild-type A. tumefaciens (Fig. S3A, P value of 0.09 by paired t test).
The impact of the dcpA mutations on biofilm formation, Congo red staining, and c-di-GMP levels in the wild-type strain was determined. Providing the plasmid-borne dcpA gene in the wild-type background depressed levels of biofilm formation (Fig. 2A, P value of <0.05 by paired t test compared to the WT value). Slight, nonsignificant decreases in Congo red staining and c-di-GMP concentrations (P value of 0.12 by paired t test compared to the WT value) were also observed (Fig. 2B and C). Introduction of the dcpA GGDEF motif (E308A) mutant plasmid (DGC−PDE+) resulted in a similar decrease (P value of 0.05 by paired t test compared to the WT value), but expression of the EAL mutant (E431A) resulted in a striking increase (P value of <0.05 by paired t test compared to the WT value) in the above phenotypes (Fig. 2A to C), suggesting that DcpA exhibits DGC enzymatic activity when PDE activity is abrogated. The concentration of c-di-GMP in the strain expressing the dcpA double mutant (E308A E431A) plasmid was not different from the plasmid-free strain (P value of 0.29 by paired t test compared to the WT value).
FIG 2 .

Genetic analysis reveals DGC activity of DcpA. (A) Biofilms grown for 48 h on PVC coverslips were quantified as described in the legend to Fig. 1C. A wild-type strain with no plasmid inserted (-) is shown. The error bars show 1 standard deviation (SD). (B) Congo red colony phenotypes of A. tumefaciens strains after 48 h of growth at 28°C (vertically aligned with strain designations in panel A). (C) Quantification of intracellular levels of c-di-GMP in the indicated A. tumefaciens or E. coli strains. A DcpA variant containing catalytic site GGDEF→GGDAF mutation (DGC−) and a DcpA variant containing catalytic site EAL→AAL mutation (PDE−) were tested. c-di-GMP was measured by LC-MS/MS as described in Text S1 in the supplemental material. Values are results of two (A. tumefaciens) or three (E. coli) independent biological replicates consisting of three technical replicates each. The error bars show 1 SD.
DcpA exhibits DGC activity in a heterologous host.
Production of c-di-GMP is often controlled by a variety of upstream regulatory pathways, specific to the bacterial species in which they have evolved (9, 10). We hypothesized that A. tumefaciens might express regulatory proteins that modulate the DGC and PDE activity of DcpA. To examine enzymatic activity in a heterologous host lacking any A. tumefaciens-specific regulatory machinery, wild-type and mutant variants of DcpA were expressed from a multicopy plasmid in Escherichia coli DH5α. E. coli DH5α has a low basal level of c-di-GMP, and we have previously used it as an assay strain to show that several A. tumefaciens DGCs can elevate intracellular c-di-GMP levels significantly above the low endogenous E. coli background (50).
Unlike wild-type A. tumefaciens, expression of plasmid-borne wild-type dcpA in E. coli increased intracellular c-di-GMP concentrations by more than 2 orders of magnitude compared to the plasmid-less background (Fig. 2C, P value of <0.05 by paired t test). This increase in c-di-GMP was entirely dependent on the DGC activity of DcpA, as the GGDEF (E308A) mutation did not increase or decrease c-di-GMP levels compared to the wild type (P value of 0.26 by paired t test). This plasmid derivative is not generally dysfunctional, as it was able to successfully complement the A. tumefaciens ΔdcpA mutant for its presumptive PDE activity (Fig. 1A). The DGC activity of the EAL (E431A) mutant was similar to that of wild-type DcpA, indicating that the DGC activity of DcpA is functionally independent of PDE activity when the protein is expressed in E. coli. This is consistent with our finding in A. tumefaciens in which the PDE− mutant exhibited strong DGC activity (Fig. 1A and 2C). Expression of the double mutant (E308A E431A) had no effect on c-di-GMP levels. Overall, these data suggest that DcpA acts as a potent DGC when expressed in E. coli and that DcpA DGC activity is likely to be regulated by factors or conditions native to A. tumefaciens.
A putative pteridine reductase negatively regulates biofilm formation and UPP production in A. tumefaciens.
We had identified additional candidate UPP and attachment regulators in our prior transposon mutant screen for UPP dysregulation (50) and hypothesized that some of these might consolidate into common control pathways. Of the four classes of candidate regulators, transposon mutations in Atu1130 exhibited the highest level of biofilm formation when disrupted (50). As detailed below, Atu1130 and DcpA are functionally linked in their control over UPP production and surface attachment in general.
The Atu1130 gene is on the circular chromosome, and it is expressed divergently from an outer membrane protein homolog designated aopB (Atu1131). The 3′ end of the Atu1130 coding sequence overlaps by 4 bp with the downstream gene homologous to the UvrC UV repair exinuclease protein (Atu1129), suggesting that these genes are translationally coupled. The Atu1130 gene encodes a 262-aa gene product that shares significant sequence similarity with pteridine reductase proteins, a subclass of the short-chain dehydrogenase/reductase protein family (53, 54). The annotated pteridine reductase domain extends from positions 14 to 247 and is the only recognized domain of the Atu1130 protein. Due to these characteristics, Atu1130 was renamed PruA (pteridine reductase regulator of UPP A). Pteridine reductases catalyze the NADPH-dependent reduction of metabolites derived from GTP, known as pterins, to their biologically active forms. The most well-known pterin-containing molecules are folates, while other pterin compounds, such as the molybdopterin cofactor, biopterins, and monapterins, function as enzymatic prosthetic groups that drive redox catalysis (55–58). The pteridine reductase family is characterized by a conserved YXXXK catalytic motif that is necessary for enzymatic function (53, 59, 60). PruA contains the canonical YXXXK motif (see Fig. S4A in the supplemental material) and exhibits predicted secondary and tertiary structural homology with well-characterized pteridine reductases such as PTR1 from Leishmania donovani (Fig. S4D).
An in-frame deletion of pruA that retains the potential translational coupling with uvrC results in an approximately fivefold elevation of biofilm formation compared to the wild type (Fig. 3A, P value of <0.05 by paired t test). As predicted by elevated biofilm levels, a pruA mutant also had slight, but statistically higher levels of c-di-GMP than the wild type did (see Fig. S3 in the supplemental material, P value of <0.05 by paired t test). The ΔpruA mutant also formed dark red colonies on Congo red-containing medium (Fig. 3B), indicative of elevated levels of cellulose and UPP. The elevated levels of biofilm formation and Congo red staining were returned to normal levels (Fig. 3A and B) by ectopic expression of a plasmid-borne copy of pruA (Plac-pruA), confirming that the mutant phenotypes are not due to polarity on uvrC.
FIG 3 .

Enzymatic activity of PruA required for control of attachment. (A) Biofilms grown on PVC coverslips for 48 h were quantified as described in the legend to Fig. 1C. A wild-type strain with no plasmid inserted (-) and the pruA mutant strain with no plasmid inserted (-) are indicated. The error bars show 1 SD. (B) Congo red colony phenotypes of the indicated A. tumefaciens strains. The bacteria were grown for 48 h at 28°C. (C) Unipolar polysaccharide (UPP) production (red) visualized by staining with Alexa Fluor 594-labeled wheat germ agglutinin (afWGA). Exponential-phase planktonic cultures were incubated with afWGA (10 µg/ml) and spotted (1 µl) onto 1% agarose pad. Bacteria were viewed at a magnification of ×100 on a Nikon E800 epifluorescence microscope (excitation, 510 to 560 nm; emission, >610 nm). The images shown are overlays of phase-contrast and fluorescence microscopy images. The images were exposed for 40 and 400 ms for phase-contrast and fluorescence microscopy, respectively. IPTG (400 µM) was added to each culture. Bars, 5 µm.
To observe UPP production directly, cells were stained using wheat germ agglutinin (WGA) conjugated to the Alexa Fluor 594 fluorescent label (af-WGA). WGA specifically binds to N-acetylglucosamine-containing polysaccharides and specifically labels the site of UPP production in A. tumefaciens (50, 61). UPP production is tightly dependent on surface attachment in wild-type A. tumefaciens (62), and little to no WGA staining was observed for planktonically grown cultures (Fig. 3C). In contrast, the ΔpruA mutant exhibited abundant UPP staining and high levels of cellular aggregation, also consistent with cellulose production (Fig. 3C). UPP staining was concentrated in the large cell aggregates, but it was also visible on single cells. As UPP production is normally not observed in planktonically grown cells (50, 62), synthesis of the adhesin is thus uncoupled from surface attachment in the ΔpruA mutant, consistent with other regulatory mutants identified in our initial screen such as visN and visR (50). The plasmid-borne pruA gene complements the mutant phenotypes back to wild-type levels. These observations suggest that PruA is an important regulatory player in the pathway controlling the A. tumefaciens motile-to-sessile transition.
PruA enzymatic activity required for function.
To test whether the enzymatic activity of PruA was required for negative regulation of attachment phenotypes, a site-directed mutation (Y163A) that altered the tyrosine residue in the presumptive active site (YXXXK) of PruA to an alanine was generated. In other systems, these mutations have been shown to abolish pteridine reductase activity but not affect protein stability (60). A plasmid expressing wild-type pruA can diminish biofilm formation and Congo red phenotypes in a ΔpruA mutant (Fig. 3A and B). In contrast, the PruA Y163A mutation abolished the ability of this plasmid to complement, exhibiting biofilm levels and Congo red staining indistinguishable from those of the mutant without the plasmid (Fig. 3A and B, P value of 0.34 by paired t test). The Y163A mutation was also introduced into the native genomic context of pruA by allelic replacement, and this mutant was similar to the ΔpruA deletion strain with respect to all measured phenotypes, including biofilm formation (see Fig. S5 in the supplemental material) and Congo red staining (data not shown). PruA enzymatic activity thus appears to inhibit surface attachment processes, including UPP production in A. tumefaciens.
PruA orthologs from other bacteria can rescue the A. tumefaciens pruA mutant.
PruA orthologs are observed in several related members of the Rhizobiales order and Alphaproteobacteria class, including Sinorhizobium meliloti, Caulobacter crescentus, Ruegeria pomeroyi, and Brucella abortus, with all homologs exhibiting approximately 40% amino acid identity or higher. Each of the PruA orthologs possesses a canonical YXXXK motif (see Fig. S4A in the supplemental material), suggesting a conserved enzymatic function. When expressed from Plac on a plasmid, all of these homologs drove the ΔpruA biofilm phenotype and Congo red staining toward wild-type levels (Fig. S4B and C). The closest E. coli homolog to PruA is known as FolM (26% identity to PruA), an experimentally validated pteridine reductase. FolM functions as a dihydropterin reductase, reducing dihydromonapterin (H2-MPt) to tetrahydromonapterin (H4-MPt) (58). Strikingly, ectopic expression of folM in A. tumefaciens also partially reverses the biofilm and Congo red phenotypes of the ΔpruA mutant (Fig. S4B and C).
PruA is required for production of a novel pterin.
In order to evaluate the activity of PruA, we examined the intracellular pterin profiles in fractionated preparations from A. tumefaciens using high-performance liquid chromatography (HPLC) with fluorescence detection. All analyses were performed on oxidized extracts, as pterin derivatives in their oxidized state are strongly fluorescent. This technique is highly sensitive for oxidized pterin species, but its use precludes the determination of the reduction state of the pterin. This analysis showed the presence of three major pterin-containing peaks (Fig. 4A). The peak that eluted at 11.7 min corresponds to pterin (Fig. 4B), a major degradation product of tetrahydrofolate, commonly found in cell extracts (63). The peak at 3.8 min is likely a folate biosynthetic intermediate containing a phosphate group, and although this compound appeared to decrease in the cells lacking PruA, the significance of this is not yet clear. The peak eluting at 12.5 min had a characteristic pterin absorbance spectrum and was strikingly absent from the ΔpruA and pruA Y163A mutants but was present when the ΔpruA mutant was complemented with the Plac-pruA plasmid.
FIG 4 .
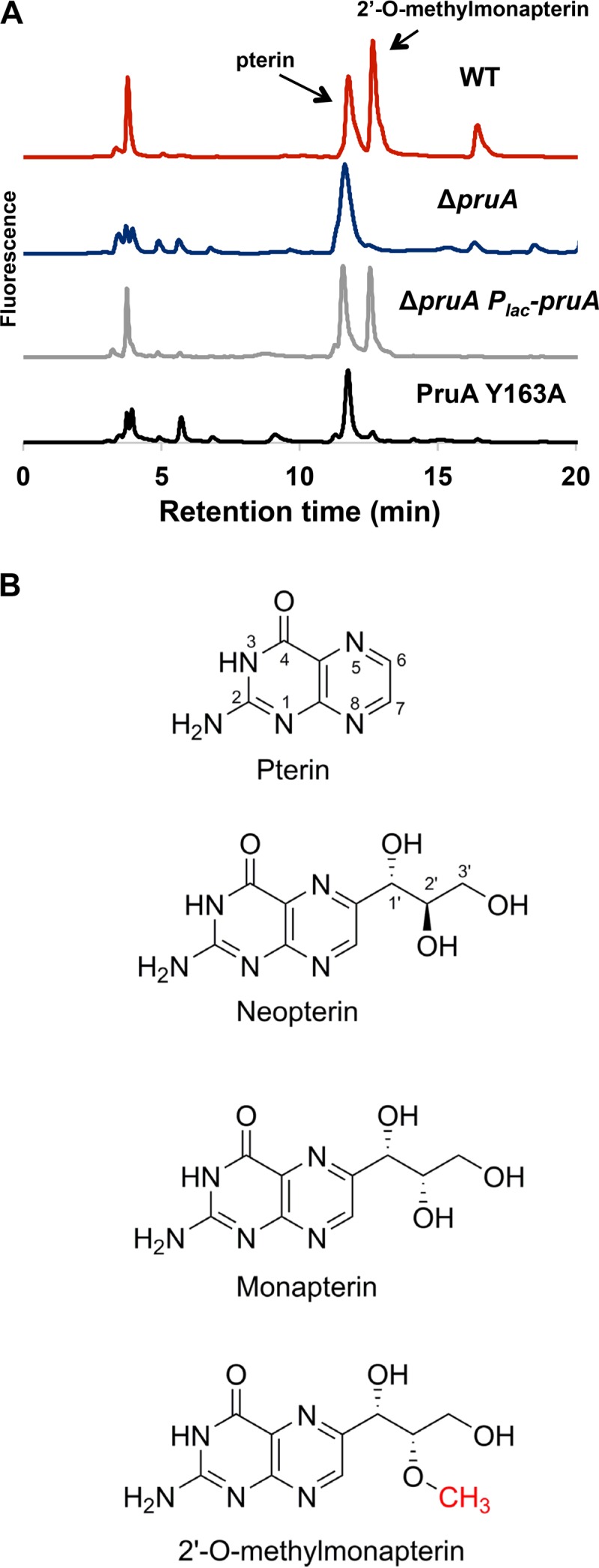
PruA enzymatic activity required for pterin synthesis. (A) HPLC traces of pterin extracts from the indicated A. tumefaciens strains. Lyophilized cells were resuspended, and pterins were extracted, purified, and analyzed by HPLC as described in Materials and Methods. (B) Pterin chemical structures (ring positions shown).
Since the species eluting at 12.5 min did not match any known pterin based on retention time, the peak was collected and analyzed further by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The unknown pterin had a (M+H)+ of 268 m/z and (M-H)− of 266 m/z, corresponding to a molecular mass of 267 Da, consistent with the addition of a methyl group to neopterin or monapterin, which differ only in stereochemistry at the 2′ position (Fig. 4B). Collision-induced dissociation (CID) of the (M-H)− precursor ion gave rise to a pterin fragment at 162 m/z, indicating that the methyl group is not on the pterin portion of the molecule and must be present on the side chain to generate an O-methylated neopterin or monapterin derivative. FolM from E. coli reduces 7,8-dihydromonapterin (H2MPt) to 5,6,7,8-tetrahydromonapterin (H4MPt) but has no activity with 7,8-dihydroneopterin (58). Given the similarity of PruA and FolM, it seemed likely that the PruA substrate is 7,8-dihydromonapterin and that the methylated pterin compound we identified in A. tumefaciens is a methylated monapterin rather than neopterin. We synthesized 6-methoxymethylpterin (lacking the full side chain of the monapterin) and analyzed the compound by LC-MS/MS. The MS/MS data obtained from this compound were very different from the data obtained from the unknown pterin isolated from A. tumefaciens (data not shown), indicating that the methyl group was not likely at the 1′ position, and further analysis led us to propose O-methylation at the 2′ position. We therefore synthesized 2′-O-methylmonapterin, and HPLC and LC-MS/MS analyses revealed that it had the same retention time and MS/MS spectrum as the A. tumefaciens pterin (Fig. S7B and S7C). The unknown oxidized pterin present in A. tumefaciens is therefore 2′-O-methylmonapterin.
A. tumefaciens PruA was purified as a hexahistidinyl-tagged PruA protein (His6-PruA) from E. coli. The purified protein was tested for enzymatic activity, monitoring oxidation of NADPH, and was found to reduce H2MPt (specific activity, 0.726 ± 0.039 µmol min−1 mg−1), but it had no activity with dihydroneopterin, dihydrofolate, or other oxidized pterins. Thus, PruA does function as predicted for a pteridine reductase. Taken together, our results indicate that PruA can generate H4MPt from H2HPt but that another cellular activity in A. tumefaciens must convert this to 2′-O-methyltetrahydromonapterin (2′-OMet-H4MPt). One or both of these pterins regulate DcpA activity and attachment.
Regulatory connection between PruA and DcpA.
In our initial transposon mutant screen (50), pruA and dcpA mutants exhibited similar phenotypes, and thus, we hypothesized that they might function together. To test the hypothesis that pruA and dcpA are part of the same regulatory pathway, plasmid-borne copies of either pruA or dcpA were introduced independently into the ΔdcpA and ΔpruA mutant backgrounds. Expressing pruA in a dcpA mutant did not change its phenotype compared to the untransformed dcpA mutant (data not shown). In contrast, expressing dcpA in a pruA background (deleted for cellulose [Cel−] to reduce excessive clumping) resulted in an approximately twofold increase above the already elevated biofilm phenotype of the ΔpruA mutant alone (Fig. 5A, P value of <0.05 by paired t test). This increased biofilm formation was accompanied by increased cell aggregation and elevated UPP staining (Fig. 5B) and an increase of more than 2 orders of magnitude in intracellular c-di-GMP concentrations compared to the ΔpruA strain (Fig. 5A, P value of <0.05 by paired t test). Introducing the Plac-dcpA plasmid into the chromosomal pruA catalytic site mutant (Y163A) increased biofilm formation similar to that observed in the ΔpruA background (see Fig. S5 in the supplemental material). Thus, in contrast to the PDE activity of DcpA that predominates in wild-type A. tumefaciens, DcpA acts as a strong DGC in the absence of an enzymatically active copy of pruA, analogous to its activity when expressed in E. coli.
FIG 5 .

Regulatory connections between PruA and DcpA. (A) Biofilms grown for 48 h on PVC coverslips (gray bars) were quantified as described in the legend to Fig. 1C. The error bars show 1 SD. c-di-GMP levels were also quantified (black bars). Strains were grown to stationary phase, nucleotides were extracted, and c-di-GMP was measured as described in Materials and Methods. A DcpA variant with catalytic site GGDEF→GGDAF mutation (DGC−), DcpA variant containing catalytic site EAL->AAL mutation (PDE−), and mutant with no plasmid inserted (-) were used. The Δcel background was utilized to reduce excess clumping. Values are results of three (biofilm) or two (c-di-GMP) independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. (B) UPP production visualized by staining with afWGA, as described in the legend to Fig. 3C. IPTG (400 µM) was added to each culture. Bars, 5 µm. (C) Adherent biomass grown for 48 h was quantified as described above for panel A, with IPTG omitted.
The plasmid carrying the GGDEF mutant allele (E308A) of dcpA was introduced into the ΔpruA background, and it did not stimulate elevated biofilm formation or increase c-di-GMP levels (Fig. 5A, P value of >0.05 by paired t test). The elevation of biofilm and c-di-GMP levels was still strongly stimulated by introduction of the plasmid-borne EAL mutant (E431A) of dcpA in the ΔpruA background, suggesting again that DcpA DGC activity is functionally independent of an enzymatically active PDE domain. Plasmid-borne expression of the GGDEF-EAL double mutant (E308A E431A) had no effect on the ΔpruA phenotypes. It seemed likely that the ΔpruA phenotypes might be DcpA dependent, and a ΔpruA ΔdcpA double mutant was found to diminish biofilm levels below that of the ΔpruA single mutant to that of the ΔdcpA mutant (Fig. 5C, P value of <0.05 by paired t test). Thus, the dcpA mutation is epistatic to pruA and is downstream in the regulatory pathway. The very strong pruA mutant phenotypes are also likely due to the combined effect of a decrease in DcpA PDE activityand sustained or stimulated DcpA DGC activity.
PruA-dependent regulation of DcpA requires a putative pterin-binding protein.
Despite our observations for PruA and DcpA, it was not clear how PruA-dependent pterin production could control DcpA. As mentioned above, dcpA is immediately preceded by Atu3496 in a putative operon (Fig. 1A) (50). In fact, several transposon insertion mutations were identified in the Atu3496 gene during the initial mutant screen. Atu3496 is a 169-aa protein, the majority of which (positions 34 to 172) shows modest but significant similarity to an oxidoreductase molybdopterin-binding domain (pfam00174, BLAST E value of 3.14 × 10−8, specific match to E. coli YedY, HHPred E value of 1.5 × 10−31). These domains are generally utilized for molybdopterin cofactor binding, through conjugation at a conserved cysteine residue that is required for the proper function of redox enzymes such as sulfite oxidases (55). This cysteine residue is absent in the Atu3496 gene, and several conserved residues are divergent, suggesting that it may not bind molybdopterin specifically. There are several homologous proteins in other Alphaproteobacteria that also lack this conserved cysteine (see Fig. S6A in the supplemental material). We hypothesized that this domain in Atu3496 might instead provide response to the pterin derivative produced by PruA. We tentatively designated the gene pruR (pteridine reductase regulator of UPP receptor).
An in-frame deletion in pruR was generated, with care taken not to disrupt the start site or ribosome-binding site of dcpA. Deletion of pruR resulted in an approximately 3.5-fold increase in biofilm formation compared to the wild type (Fig. 6A, P value of <0.05 by paired t test), increased af-WGA labeling (Fig. 6B), and elevated Congo red staining (data not shown), indicating that PruR negatively regulates attachment processes in A. tumefaciens. Ectopic expression of a plasmid-borne pruR gene reduced the biofilm levels of a ΔpruR mutant only slightly (P value of <0.05 by paired t test), whereas providing expression of a plasmid with both pruR and dcpA in their normal tandem configuration reduced biofilm formation very strongly to below wild-type levels (Fig. 6A, P value of <0.05 by paired t test). The strong suppressive effect on the pruR phenotype observed with the Plac-pruR-dcpA tandem expression plasmid was abolished in a ΔpruA ΔpruR double mutant (see Fig. S6B in the supplemental material). The requirement for this dual-gene plasmid suggests a strong cis association and perhaps a cotranslational interaction. We are confident that the ΔpruR mutation does not disrupt the downstream dcpA, and this is further supported in that providing a plasmid-borne dcpA alone in this mutant stimulates biofilm formation rather than returning the elevated levels of pruR closer to the wild-type levels, the opposite effect one would predict for correcting a dcpA deficiency (see below). These data suggest that pruR and pruA might act in concert to regulate DcpA activity
FIG 6 .

PruR negatively regulates biofilm formation. (A) Biofilms grown for 48 h on PVC coverslips were quantified as described in the legend to Fig. 1C. A ΔpruR mutant with no plasmid inserted is indicated (-). The error bars show 1 SD. (B) UPP production visualized by staining with afWGA, as described in the legend to Fig. 3C. Bars, 5 µm. (C) Adherent biomass grown for 48 h was quantified as described above for panel A, with IPTG omitted. The Δcel background was utilized to reduce excess clumping. (D) Quantification of c-di-GMP levels in the indicated E. coli strains. The E. coli strains were grown to late exponential phase, and nucleotides were extracted and quantified as described in Text S1 in the supplemental material. Values are results of two independent biological replicates each with three technical replicates. The error bars show 1 SD.
PruR is required to maintain the dominant PDE activity of DcpA.
We predicted that PruR would regulate the dual enzymatic activities of DcpA, and thus, a switch in DcpA enzymatic activity might be observed in a pruR mutant similar to pruA mutants. To test this, the Plac-dcpA plasmid was expressed in the A. tumefaciens ΔpruR mutant (also mutated for the cellulose biosynthetic genes to prevent aggregation) and assayed for biofilm formation. As observed before, a ΔpruR mutant exhibited higher biofilm levels than the wild type did (Fig. 6C, P value of <0.05 by paired t test). Expression of the plasmid-borne dcpA increased biofilm formation approximately fourfold above the ΔpruR mutant (Fig. 6C, P value of <0.05 by paired t test). The dcpA-mediated increase in biofilm formation was abrogated in the DcpA (E308A) GGDEF catalytic site mutant (Fig. 6C). However, plasmid-borne expression of the DcpA (E431A) EAL catalytic site mutant stimulated biofilm formation equivalent to the wild-type gene, indicating again that the DGC activity was retained in the PDE mutant (Fig. 6C). Furthermore, introduction of the Plac-pruR-dcpA plasmid into E. coli did not increase c-di-GMP levels, in contrast to the dramatic increases observed with the Plac-dcpA plasmid (Fig. 6D). Overall, these data suggest that PruR, like PruA, controls the enzymatic activity of DcpA. PruR and PruA by themselves are both necessary but not sufficient for proper regulation of DcpA, as DcpA functions as a PDE only in the presence of both PruA and PruR.
DISCUSSION
In this study, we confirm and expand our initial report (50) on dcpA (Atu3495) in A. tumefaciens. We demonstrate that DcpA PDE activity, and specifically the EAL domain, is essential for the negative regulation of UPP adhesin production, cellulose synthesis, and biofilm formation, through its impact on c-di-GMP levels that regulate production of these polysaccharides and other aspects of the motile-to-sessile transition. We also describe, through the use of site-specific mutagenesis, the ability of DcpA to exhibit in vivo DGC activity, and that the DGC and PDE activities are independent of each other. Our findings reveal that the DcpA activity is controlled through a novel, pterin-dependent regulatory mechanism.
A dual-function diguanylate cyclase-phosphodiesterase in A. tumefaciens.
Demonstration of both DGC and PDE enzymatic activities from the same protein is not typical, as a large percentage of proteins with both GGDEF and EAL domains exhibit only one of the two functionalities (30, 31). For those examples of proteins with both activities, the overarching regulatory networks that control them are often unclear, and in vivo data are scarce. We find here that the enzymatic activity of PruA (Atu1130), is responsible for the negative control of attachment phenotypes through the PDE activity of DcpA, as manifested through the function of the PruR protein (Atu3496).
There are a limited number of well-studied dual-function proteins with both PDE and DGC activity. Arguably the best-known DGC-PDE protein is ScrC from Vibrio parahaemolyticus, which positively regulates swarming and negatively regulates capsular polysaccharide (CPS) production (37). ScrC is a transmembrane protein with a periplasmic domain, and its PDE activity requires ScrB, a periplasmic solute-binding protein homolog, and ScrA, homologous to pyridoxal-dependent-enzymes (36). ScrA generates an extracellular signal (S-signal), and together with ScrB, it promotes the PDE activity of ScrC. In the absence of ScrA and ScrB, the DGC activity of ScrC dominates. Thus, for both ScrC and DcpA, c-di-GMP turnover is controlled by two upstream regulatory partners (ScrA and ScrB in V. parahaemolyticus and PruA and PruR in A. tumefaciens). In each case, one regulatory partner is required to synthesize a small molecule, S-signal by ScrA and a pterin by PruA. Another recently described dual-function DGC-PDE is BphG1 of Rhodobacter sphaeroides, consisting of an N-terminal photosensory module coupled to a C-terminal output module containing GGDEF and EAL domains (34). Purified full-length BphG1 demonstrates PDE activity in vitro, but ablation of the EAL domain results in strong DGC activity. It remains unclear whether similar cleavage of the EAL domain occurs in vivo, and BphG1 DGC activity has not been observed in a native background. In contrast, for DcpA, we observe both DGC and PDE activity in vivo, and these activities have clear cellular phenotypes linked to DcpA output.
Pterin synthesis and DcpA regulation.
It is now clear that the enzymatic activity of PruA is required for DcpA PDE activity and that PruA directs the formation of 5,6,7,8-tetrahydromonapterin (H4MPt). Extracts from A. tumefaciens reveal PruA-dependent production of the novel monapterin 2′-OMet-H4MPt, and thus, the H4MPt that is the direct product of PruA activity is likely to be a substrate for methylation by an as-yet-unidentified enzyme (Fig. 7; see Fig. S7A in the supplemental material). Since pterins readily undergo oxidation and degradation upon isolation from cells, we do not observe the tetrahydro product in A. tumefaciens extracts but instead observe the oxidized form of the pterin, 2′-OMet-MPt (Fig. 4A).
FIG 7 .

Model of pterin-dependent DcpA regulation. A tentative model for DcpA regulation is shown. The model is based on the findings reported here, with details provided in the text. Solid black arrows are enzymatic reactions, dashed arrows are regulatory interactions, and the squiggly arrow is a speculative environmental influence on the putative PruR-pterin (Pt) complex. Red text indicates essential genes in A. tumefaciens. IM, inner membrane; OM, outer membrane; GTs, glycosyl transferases; MoPT, molybdopterin; P-ase, phosphatase.
In well-studied systems, monapterin biosynthesis branches from folate metabolism (58), and we predict that this is similar in A. tumefaciens (Fig. 7). Both folate and monapterin are derived from GTP through the common intermediate 2,3-dihydroneopterin triphosphate (H2NPt-P3) by a GTP cyclohydrolase, encoded by folE in E. coli, and homologous to Atu1749 in A. tumefaciens (64). The well-studied molybdopterin cofactor is synthesized via a different pathway that also utilizes GTP as an intermediate (65). Analogous to the E. coli pathway for monapterin biosynthesis, the likely pathway in A. tumefaciens would share several key enzymes with folate biosynthesis, acting on either a neopterin for folate or a monapterin (Fig. 7). H2MPt formed by this pathway could then be reduced to H4-MPt. In A. tumefaciens, all of the enzymes predicted to be required prior to the PruA-dependent reaction are essential for growth (Fig. 7, red Atu genes), even in rich medium (66), thus explaining why no other transposon mutants with pruA-type phenotypes were obtained for this pathway. This also suggests that in A. tumefaciens, folate is essential for growth, but monapterin is not.
PruR-pterin interactions.
PruR is required for the PruA-dependent control of the enzymatic activity of DcpA by promoting its PDE activity. Although PruR is annotated as a molybdopterin-binding protein, it is degenerate and most importantly missing the key cysteine ligand for molybdopterin (67). As it is clear that PruA drives H4MPt synthesis, the simplest model is that PruR can respond to a monapterin. Cells with active PruA produce 2′-OMet-H4MPt, presumably derived from methylation of H4MPt, but no mutants with the pruA phenotype were obtained for any annotated methyltransferase. Therefore, it is possible that H4MPt produced by PruA is in fact the active molecule for PruR-dependent control or that the methyltransferase gene is another essential gene.
A relatively simple model is that a PruA-dependent pterin interacts with PruR, perhaps by forming a complex. Such a PruR-pterin complex could directly or indirectly interact with DcpA to promote its PDE activity (Fig. 7). More-complex models might also be envisioned, such as a mechanism by which the monapterin species limits the action of an inhibitor of PruR. Such a mechanism would be consistent with the observation that PruR effectively switches DcpA from its DGC dominant state in E. coli, a host that does not produce the novel monapterin we have detected in A. tumefaciens.
Since the PDE activity predominates under standard culture conditions in wild-type A. tumefaciens, it is possible that the PDE domain, when enzymatically active, exerts a negative regulation on the DGC domain. In ScrC of V. parahaemolyticus, the GGDEF catalytic motif is required for its PDE activity (38), but in contrast, the DcpA PDE activity appears to remain functional in GGDEF catalytic site mutants. Alternatively, both domains may be active, but the PDE activity greatly exceeds the DGC activity, leading to predominant c-di-GMP inactivation, and thus inhibition of attachment functions such as UPP and cellulose.
Pterin-dependent regulation.
It is not obvious why or how monapterins would control DcpA activity. The simplest explanation is that the intracellular concentration of a specific monapterin(s) directly modulates DcpA, probably through interactions with PruR. The monapterin pathway derives from GTP and folate synthesis, and thus, the concentration of these compounds could reflect the overall levels of GTP. Alternatively, the monapterin could function as several other GTP derivatives such as c-di-GMP and ppGpp (10, 68, 69) directly as a second messenger.
It is also plausible that monapterin might function as a sensor of the cytoplasmic redox state through association with PruR and perhaps DcpA. PAS domains containing heme cofactors have been shown to modulate PDE activity through reversible binding of oxygen (70, 71). Redox sensing by flavin cofactors can regulate the enzymatic activity of a DGC protein in Acetobacter xylinum (72), and it is possible that pterins could operate in a similar capacity (58). Reduced pterins are known to act as robust antioxidants in mammalian cells (73), and high levels of oxidized neopterin are associated with increased concentrations of reactive oxygen species (ROS) (74). Additionally, several pterins are involved in regulating a redox-sensitive transcription factor in mammalian cells (75, 76). Oxygen availability has been postulated to play an important role during biofilm formation in A. tumefaciens (43), and biofilms often generate oxygen gradients (77). Sensing oxygen or changes in intracellular redox balance during the transition from the motile to sessile state could be mediated in part through monapterin control of DcpA.
The mechanism by which PruR directs the monapterin-mediated control of DcpA activity also remains to be established. An intimate relationship between PruR and DcpA is suggested by their transcriptional linkage within the same operon and the observation that their coexpression appears to potentiate the PruR-dependent stimulation of PDE activity. Regulation of DcpA via PruR could occur via several possible mechanisms, including direct binding and occlusion of the DGC domain, direct promotion of the PDE domain, or both, as well as control of proper DcpA subcellular localization. The interaction between PruR and DcpA may be through formation of a stable complex between the two proteins and a monapterin prosthetic group. Other more-complex possibilities are also possible. For example, PruR might have an as-yet-undiscovered enzymatic capability that is active only in the presence of a specific pterin cofactor, as with pterin-dependent enzymes, including phenylalanine hydroxylase (58). The enzymatic activity of PruR could directly modify DcpA or synthesize another small molecule that in turn controls DcpA activity.
Ultimately, a more detailed understanding of the PruA-PruR-DcpA pathway will result from biochemical analysis of the different regulatory components and investigation into the cellular context of its activity. Our current findings provide the first example of pterins participating in a prokaryotic signaling cascade, in this case regulating surface attachment through adhesin production in A. tumefaciens. Pterins are produced by diverse bacteria, and thus, it is possible that similar pterin-dependent control is widespread.
MATERIALS AND METHODS
Reagents, media, strains, and growth conditions.
All strains and plasmids and all oligonucleotides used in this study are provided in Tables S1 and S2 in the supplemental material, respectively. The design and verification of plasmids and engineered genetic derivatives, including site-specific mutagenesis, deletions, and allelic exchanges were performed as described in previous publications (45, 50) with specific details included in Text S1 in the supplemental material.
Biofilm analysis and UPP adhesin assays.
Biofilms of A. tumefaciens derivatives were cultivated in minimal medium on polyvinyl chloride (PVC) coverslips in 12-well plates, and adherent biomass was measured using crystal violet as in our prior publications (45, 78). Cells producing the UPP adhesin were examined using epifluorescence microscopy and the fluorescently labeled lectin af-WGA as described previously (50).
c-di-GMP measurements.
Extracts were prepared from cultures of A. tumefaciens and E. coli derivatives and analyzed using LC-MS/MS on a Quattro Premier XE mass spectrometer (Waters Corporation) coupled with an Acquity ultraperformance LC system (Waters Corporation) as described in a previous publication (50).
Extraction and analysis of pterins from A. tumefaciens.
Whole-cell extracts prepared from late-exponential-phase cultures of A. tumefaciens were prepared, and pterins were enriched using cation exchange chromatography. These preparations were analyzed using HPLC on a reverse-phase C18 column with fluorescence detection as previously described (79) and provided in Text S1 in the supplemental material in more detail.
Purification and enzymatic assay of His6-PruA.
An N-terminal hexahistidinyl-tagged PruA (His6-PruA) derivative was expressed in E. coli and affinity purified. This preparation was tested for enzymatic activity using an H2MPt substrate prepared by reduction of monapterin and a dihydrofolate substrate using an assay monitoring consumption of NADPH in the reaction spectrophotometrically using extinction coefficients for the NADPH-pterin oxidation-reduction pair.
Additional details on the materials and methods used in this study are provided in Text S1 in the supplemental material.
SUPPLEMENTAL MATERIAL
DcpA domain alignments. Alignments performed using ClustalX multiple alignment mode and neighbor-joining (NJ) clustering algorithm. (A) Alignment of the DGC domain of DcpA to characterized DGC proteins. Residues important for catalytic function are outlined in red. (B) Alignment of the DcpA EAL domain to characterized proteins containing EAL domains. Residues important for catalytic function are outlined in red. Download
Additional ΔdcpA phenotypes. (A) Congo red phenotypes of the indicated A. tumefaciens strains. Exponential-phase cultures were normalized to an OD600 of 0.5, and 5 µl of the corresponding culture was spotted onto ATGN minimal medium plates containing 100 µg/ml Congo red and 400 µM IPTG. Pictures were taken after 48 h of growth at 28°C. DGC−, DcpA variant containing catalytic site GGDEF→GGDAF mutation; PDE−, DcpA variant containing catalytic site EAL→AAL mutation. (B) Adherent bacterial biomass grown for 48 h on PVC coverslips quantified by the amount of solubilized crystal violet (CV) at A600, as described in the legend to Fig. 1C. In parallel, the OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the A600/OD600 ratio. The indicated amounts of IPTG were added. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
Intracellular c-di-GMP levels. (A) Quantification of intracellular levels of c-di-GMP in the indicated A. tumefaciens strains. Strains were grown to stationary phase, nucleotides were extracted, and measurements of c-di-GMP were obtained by LC-MS/MS as described in Materials and Methods. All strains were constructed in an extracellular polymeric substance (EPS)-negative strain background (45) in which the genes coding for the polysaccharides succinoglycan, cellulose, cyclic β-1-2 glucans, and curdlan (β-1-3-glucans) have been deleted. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
PruA orthologs complement ΔpruA phenotypes. (A) A. tumefaciens pruA aligned to orthologs from other Rhizobiales and alphaproteobacterial species. Orthologs from C. crescentus CC0417, R. pomeroyi Spo3638, S. meliloti SMc00603, Brucella melitensis bv. abortus BAB1_0721, and E. coli folM were used. Residues important for catalytic function are outlined in red. Alignments were performed using ClustalX multiple alignment mode and NJ clustering algorithm. (B) Adherent bacterial biomass grown for 48 h on PVC coverslips was quantified by the amount of solubilized crystal violet (CV) at A600 as described in the legend to Fig. 1C. In parallel, OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the A600/OD600 ratio. A mutant with no plasmid inserted is indicated (hyphen). Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. (C) Congo red phenotypes of the indicated A. tumefaciens strains. Cultures were normalized to an OD600 of 0.5, and 5 µl of corresponding culture was spotted onto ATGN minimal medium plates containing 100 µg/ml Congo red and 400 µM IPTG. The images were taken after 48 h of growth at 28°C. (D) Predicted PruA protein structure. Structure predicted using PHYRE database (http://www.sbg.bio.ic.ac.uk/phyre2). Leishmania pteridine reductase structure (PDB identifier [ID] 2XOX) is shown for comparison. Alpha helices and beta sheets are highlighted in green and red, respectively. The PruA catalytic tyrosine residue is indicated by a white arrow. Download
PruA enzymatic function necessary for regulation of DcpA DGC/PDE activity. Adherent bacterial biomass grown for 48 h on PVC coverslips quantified by the amount of solubilized crystal violet (CV) at A600 as described in the legend to Fig. 1C. In parallel, the OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the A600/OD600 ratio. IPTG (400 µM) was added to each strain. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
PruR sequence and role in attachment control. (A) Several PruR homologs are missing the cysteine required for molybdopterin binding. A. tumefaciens pruR homologs R. pomeroyi Spo0162, R. capsulatus RCAP rcc01507, R. sphaeroides RSP_0086 were aligned to yedY, a known molybdopterin-binding protein from E. coli. The canonical cysteine is highlighted in red. OR, oxidoreductase; SO, sulfite oxidase. (B) PruA and DcpA are both required for effective PruR complementation. Adherent bacterial biomass grown for 48 h on PVC coverslips quantified by the amount of solubilized crystal violet (CV) at A600 as described in the legend to Fig. 1C. In parallel, the OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the ratio A600/OD600 ratio. IPTG (400 µM) was added to each strain. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
2′-O-methyl-5,6,7,8-tetrahydromonapterin biosynthesis and structure. (A) PruA, which does not possess a predicted methyltransferase capability, reduces 7,8-dihydromonapterin to 5,6,7,8-tetrahydromonapterin, which is then methylated by an unknown methyltransferase to generate 2′-O-methyl-5,6,7,8-tetrahydromonapterin. (B) Major fragment ions derived from chemically synthesized 2′-methoxymonapterin during MS/MS analysis. Collisionally induced fragments arising from 2′-O-methylmonapterin observed in both the pterin isolated from A. tumefaciens and the chemically synthesized standard. The 191 m/z fragment was not observed upon analysis of chemically synthesized 3′-O-methylmonapterin (data not shown), providing evidence that the methylation is at the 2′ position in the new pterin from A. tumefaciens. (C) 2′-O-methylmonapterin mass spectra. MS/MS spectrum of the unknown pterin from wild-type A. tumefaciens cells (top) and of chemically synthesized 2′-O-methylmonapterin (bottom). Download
Strains and plasmids.
Oligonucleotides.
Supplemental Materials and Methods. Download
ACKNOWLEDGMENTS
This project was supported by National Institutes of Health (NIH) grant GM080546 (C.F.) and National Science Foundation grants MCB-1253684 (C.M.W.) and MCB-0722787 (R.H.W.). N.F. was funded by the Indiana University Genetics, Molecular and Cellular Sciences NIH training grant T32-GM007757.
We thank Jason Heindl for helping to edit the manuscript and the MSU Mass Spectrometry Facility for assistance with the mass spectrometry measurements.
Footnotes
Citation Feirer N, Xu J, Allen KD, Koestler BJ, Bruger EL, Waters CM, White RH, Fuqua C. 2015. A pterin-dependent signaling pathway regulates a dual-function diguanylate cyclase-phosphodiesterase controlling surface attachment in Agrobacterium tumefaciens. mBio 6(4):e00156-15. doi:10.1128/mBio.00156-15.
Contributor Information
Matthew R. Parsek, University of Washington.
E. Peter Greenberg, University of Washington.
REFERENCES
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.Danhorn T, Fuqua C. 2007. Biofilm formation by plant-associated bacteria. Annu Rev Microbiol 61:401–422. doi: 10.1146/annurev.micro.61.080706.093316. [DOI] [PubMed] [Google Scholar]
- 3.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat Rev Microbiol 8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 4.Mah TF, O’Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 5.Anderl JN, Franklin MJ, Stewart PS. 2000. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob Agents Chemother 44:1818–1824. doi: 10.1128/AAC.44.7.1818-1824.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKew BA, Taylor JD, McGenity TJ, Underwood GJ. 2011. Resistance and resilience of benthic biofilm communities from a temperate salt marsh to desiccation and rewetting. ISME J 5:30–41. doi: 10.1038/ismej.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houry A, Gohar M, Deschamps J, Tischenko E, Aymerich S, Gruss A, Briandet R. 2012. Bacterial swimmers that infiltrate and take over the biofilm matrix. Proc Natl Acad Sci U S A 109:13088–13093. doi: 10.1073/pnas.1200791109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.del Pozo JL, Patel R. 2007. The challenge of treating biofilm-associated bacterial infections. Clin Pharmacol Ther 82:204–209. doi: 10.1038/sj.clpt.6100247. [DOI] [PubMed] [Google Scholar]
- 9.Hengge R. 2009. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 10.Römling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinberger-Ohana P, Mayer R, Braun S, de Vroom E, van der Marel GA, van Boom JH, Benziman M. 1987. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature 325:279–281. doi: 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- 12.Pérez-Mendoza D, Coulthurst SJ, Humphris S, Campbell E, Welch M, Toth IK, Salmond GP. 2011. A multi-repeat adhesin of the phytopathogen, Pectobacterium atrosepticum, is secreted by a type I pathway and is subject to complex regulation involving a non-canonical diguanylate cyclase. Mol Microbiol 82:719–733. doi: 10.1111/j.1365-2958.2011.07849.x. [DOI] [PubMed] [Google Scholar]
- 13.Amikam D, Benziman M. 1989. Cyclic diguanylic acid and cellulose synthesis in Agrobacterium tumefaciens. J Bacteriol 171:6649–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HS, Gu F, Ching SM, Lam Y, Chua KL. 2010. CdpA is a Burkholderia pseudomallei cyclic di-GMP phosphodiesterase involved in autoaggregation, flagellum synthesis, motility, biofilm formation, cell invasion, and cytotoxicity. Infect Immun 78:1832–1840. doi: 10.1128/IAI.00446-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steiner S, Lori C, Boehm A, Jenal U. 2013. Allosteric activation of exopolysaccharide synthesis through cyclic di-GMP-stimulated protein-protein interaction. EMBO J 32:354–368. doi: 10.1038/emboj.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuckerman JR, Gonzalez G, Sousa EHS, Wan X, Saito JA, Alam M, Gilles-Gonzales M-A. 2009. An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry 48:9764–9774. doi: 10.1021/bi901409g. [DOI] [PubMed] [Google Scholar]
- 17.Boehm A, Kaiser M, Li H, Spangler C, Kasper CA, Ackermann M, Kaever V, Sourjik V, Roth V, Jenal U. 2010. Second messenger-mediated adjustment of bacterial swimming velocity. Cell 141:107–116. doi: 10.1016/j.cell.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Bobrov AG, Kirillina O, Ryjenkov DA, Waters CM, Price PA, Fetherston JD, Mack D, Goldman WE, Gomelsky M, Perry RD. 2011. Systematic analysis of cyclic di-GMP signalling enzymes and their role in biofilm formation and virulence in Yersinia pestis. Mol Microbiol 79:533–551. doi: 10.1111/j.1365-2958.2010.07470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pratt JT, Tamayo R, Tischler AD, Camilli A. 2007. PilZ domain proteins bind cyclic diguanylate and regulate diverse processes in Vibrio cholerae. J Biol Chem 282:12860–12870. doi: 10.1074/jbc.M611593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curtis PD, Brun YV. 2010. Getting in the loop: regulation of development in Caulobacter crescentus. Microbiol Mol Biol Rev 74:13–41. doi: 10.1128/MMBR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paul R, Weiser S, Amiot NC, Chan C, Schirmer T, Giese B, Jenal U. 2004. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev 18:715–727. doi: 10.1101/gad.289504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newell PD, Boyd CD, Sondermann H, O’Toole GA. 2011. A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol 9:e1000587. doi: 10.1371/journal.pbio.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan C, Paul R, Samoray D, Amiot NC, Giese B, Jenal U, Schirmer T. 2004. Structural basis of activity and allosteric control of diguanylate cyclase. Proc Natl Acad Sci U S A 101:17084–17089. doi: 10.1073/pnas.0406134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. 2008. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shikuma NJ, Fong JC, Yildiz FH. 2012. Cellular levels and binding of c-di-GMP control subcellular localization and activity of the Vibrio cholerae transcriptional regulator VpsT. PLoS Pathog 8:e1002719. doi: 10.1371/journal.ppat.1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simm R, Morr M, Kader A, Nimtz M, Römling U. 2004. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol Microbiol 53:1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- 27.Rao F, Yang Y, Qi Y, Liang ZX. 2008. Catalytic mechanism of cyclic di-GMP-specific phosphodiesterase: a study of the EAL domain-containing RocR from Pseudomonas aeruginosa. J Bacteriol 190:3622–3631. doi: 10.1128/JB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt AJ, Ryjenkov DA, Gomelsky M. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol 187:4774–4781. doi: 10.1128/JB.187.14.4774-4781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan RP, Fouhy Y, Lucey JF, Crossman LC, Spiro S, He YW, Zhang LH, Heeb S, Cámara M, Williams P, Dow JM. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc Natl Acad Sci U S A 103:6712–6717. doi: 10.1073/pnas.0600345103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Christen M, Christen B, Folcher M, Schauerte A, Jenal U. 2005. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J Biol Chem 280:30829–30837. doi: 10.1074/jbc.M504429200. [DOI] [PubMed] [Google Scholar]
- 31.Tal R, Wong HC, Calhoon R, Gelfand D, Fear AL, Volman G, Mayer R, Ross P, Amikam D, Weinhouse H, Cohen A, Sapir S, Ohana P, Benziman M. 1998. Three cdg operons control cellular turnover of cyclic di-GMP in Acetobacter xylinum: genetic organization and occurrence of conserved domains in isoenzymes. J Bacteriol 180:4416–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferreira RB, Chodur DM, Antunes LC, Trimble MJ, McCarter LL. 2012. Output targets and transcriptional regulation by a cyclic dimeric GMP-responsive circuit in the Vibrio parahaemolyticus Scr network. J Bacteriol 194:914–924. doi: 10.1128/JB.05807-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu N, Xu Y, Hossain S, Huang N, Coursolle D, Gralnick JA, Boon EM. 2012. Nitric oxide regulation of cyclic di-GMP synthesis and hydrolysis in Shewanella woodyi. Biochemistry 51:2087–2099. doi: 10.1021/bi201753f. [DOI] [PubMed] [Google Scholar]
- 34.Tarutina M, Ryjenkov DA, Gomelsky M. 2006. An unorthodox bacteriophytochrome from Rhodobacter sphaeroides involved in turnover of the second messenger c-di-GMP. J Biol Chem 281:34751–34758. doi: 10.1074/jbc.M604819200. [DOI] [PubMed] [Google Scholar]
- 35.Bharati BK, Sharma IM, Kasetty S, Kumar M, Mukherjee R, Chatterji D. 2012. A full-length bifunctional protein involved in c-di-GMP turnover is required for long-term survival under nutrient starvation in Mycobacterium smegmatis. Microbiology 158:1415–1427. doi: 10.1099/mic.0.053892-0. [DOI] [PubMed] [Google Scholar]
- 36.Trimble MJ, McCarter LL. 2011. Bis-(3′-5′)-cyclic dimeric GMP-linked quorum sensing controls swarming in Vibrio parahaemolyticus. Proc Natl Acad Sci U S A 108:18079–18084. doi: 10.1073/pnas.1113790108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boles BR, McCarter LL. 2002. Vibrio parahaemolyticus scrABC, a novel operon affecting swarming and capsular polysaccharide regulation. J Bacteriol 184:5946–5954. doi: 10.1128/JB.184.21.5946-5954.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferreira RB, Antunes LC, Greenberg EP, McCarter LL. 2008. Vibrio parahaemolyticus ScrC modulates cyclic dimeric GMP regulation of gene expression relevant to growth on surfaces. J Bacteriol 190:851–860. doi: 10.1128/JB.01462-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Larebeke N, Engler G, Holsters M, Van den Elsacker S, Zaenen I, Schilperoort RA, Schell J. 1974. Large plasmid in Agrobacterium tumefaciens essential for crown gall-inducing ability. Nature 252:169–170. doi: 10.1038/252169a0. [DOI] [PubMed] [Google Scholar]
- 40.Watson B, Currier TC, Gordon MP, Chilton MD, Nester EW. 1975. Plasmid required for virulence of Agrobacterium tumefaciens. J Bacteriol 123:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Escobar MA, Dandekar AM. 2003. Agrobacterium tumefaciens as an agent of disease. Trends Plant Sci 8:380–386. doi: 10.1016/S1360-1385(03)00162-6. [DOI] [PubMed] [Google Scholar]
- 42.Abarca-Grau AM, Penyalver R, López MM, Marco-Noales E. 2011. Pathogenic and non-pathogenic Agrobacterium tumefaciens, A. rhizogenes and A. vitis strains form biofilms on abiotic as well as on root surfaces. Plant Pathol 60:416–425. doi: 10.1111/j.1365-3059.2010.02385.x. [DOI] [Google Scholar]
- 43.Ramey BE, Matthysse AG, Fuqua C. 2004. The FNR-type transcriptional regulator SinR controls maturation of Agrobacterium tumefaciens biofilms. Mol Microbiol 52:1495–1511. doi: 10.1111/j.1365-2958.2004.04079.x. [DOI] [PubMed] [Google Scholar]
- 44.Tomlinson AD, Fuqua C. 2009. Mechanisms and regulation of polar surface attachment in Agrobacterium tumefaciens. Curr Opin Microbiol 12:708–714. doi: 10.1016/j.mib.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu J, Kim J, Danhorn T, Merritt PM, Fuqua C. 2012. Phosphorus limitation increases attachment in Agrobacterium tumefaciens and reveals a conditional functional redundancy in adhesin biosynthesis. Res Microbiol 163:674–684. doi: 10.1016/j.resmic.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matthysse AG, Holmes KV, Gurlitz RH. 1981. Elaboration of cellulose fibrils by Agrobacterium tumefaciens during attachment to carrot cells. J Bacteriol 145:583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthysse AG, Marry M, Krall L, Kaye M, Ramey BE, Fuqua C, White AR. 2005. The effect of cellulose overproduction on binding and biofilm formation on roots by Agrobacterium tumefaciens. Mol Plant Microbe Interact 18:1002–1010. doi: 10.1094/MPMI-18-1002. [DOI] [PubMed] [Google Scholar]
- 48.Heindl JE, Wang Y, Heckel BC, Mohari B, Feirer N, Fuqua C. 2014. Mechanisms and regulation of surface interactions and biofilm formation in Agrobacterium. Front Plant Sci 5:176. doi: 10.3389/fpls.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barnhart DM, Su S, Baccaro BE, Banta LM, Farrand SK. 2013. CelR, an ortholog of the diguanylate cyclase PleD of Caulobacter, regulates cellulose synthesis in Agrobacterium tumefaciens. Appl Environ Microbiol 79:7188–7202. doi: 10.1128/AEM.02148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J, Kim J, Koestler BJ, Choi JH, Waters CM, Fuqua C. 2013. Genetic analysis of Agrobacterium tumefaciens unipolar polysaccharide production reveals complex integrated control of the motile-to-sessile switch. Mol Microbiol 89:929–948. doi: 10.1111/mmi.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schirmer T, Jenal U. 2009. Structural and mechanistic determinants of c-di-GMP signalling. Nat Rev Microbiol 7:724–735. doi: 10.1038/nrmicro2203. [DOI] [PubMed] [Google Scholar]
- 52.Pérez-Mendoza D, Aragón IM, Prada-Ramírez HA, Romero-Jiménez L, Ramos C, Gallegos MT, Sanjuán J. 2014. Responses to elevated c-di-GMP levels in mutualistic and pathogenic plant-interacting bacteria. PLoS One 9:e91645. doi: 10.1371/journal.pone.0091645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jörnvall H, Persson B, Krook M, Atrian S, Gonzàlez-Duarte R, Jeffery J, Ghosh D. 1995. Short-chain dehydrogenases/reductases (SDR). Biochemistry 34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 54.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jörnvall H. 2003. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact 143-144:247–253. doi: 10.1016/S0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 55.Kisker C, Schindelin H, Pacheco A, Wehbi WA, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC. 1997. Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell 91:973–983. doi: 10.1016/S0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- 56.Thöny B, Auerbach G, Blau N. 2000. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J 347:1–16. doi: 10.1042/0264-6021:3470001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gourley DG, Schüttelkopf AW, Leonard GA, Luba J, Hardy LW, Beverley SM, Hunter WN. 2001. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat Struct Biol 8:521–525. doi: 10.1038/88584. [DOI] [PubMed] [Google Scholar]
- 58.Pribat A, Blaby IK, Lara-Nunez A, Gregory JF III, de Crecy-Lagard V, Hanson AD. 2010. FolX and FolM are essential for tetrahydromonapterin synthesis in Escherichia coli and Pseudomonas aeruginosa. J Bacteriol 192:475–482. doi: 10.1128/JB.01198-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeffery J, Cummins L, Carlquist M, Jörnvall H. 1981. Properties of sorbitol dehydrogenase and characterization of a reactive cysteine residue reveal unexpected similarities to alcohol dehydrogenases. Eur J Biochem 120:229–234. doi: 10.1111/j.1432-1033.1981.tb05693.x. [DOI] [PubMed] [Google Scholar]
- 60.Ensor CM, Tai HH. 1991. Site-directed mutagenesis of the conserved tyrosine 151 of human placental NAD+-dependent 15-hydroxyprostaglandin dehydrogenase yields a catalytically inactive enzyme. Biochem Biophys Res Commun 176:840–845. doi: 10.1016/S0006-291X(05)80262-1. [DOI] [PubMed] [Google Scholar]
- 61.Kim J, Heindl JE, Fuqua C. 2013. Coordination of division and development influences complex multicellular behavior in Agrobacterium tumefaciens. PLoS One 8:e56682. doi: 10.1371/journal.pone.0056682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li G, Brown PJ, Tang JX, Xu J, Quardokus EM, Fuqua C, Brun YV. 2012. Surface contact stimulates the just-in-time deployment of bacterial adhesins. Mol Microbiol 83:41–51. doi: 10.1111/j.1365-2958.2011.07909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reed LS, Archer MC. 1980. Oxidation of tetrahydrofolic acid by air. J Agric Food Chem 28:801–805. doi: 10.1021/jf60230a044. [DOI] [Google Scholar]
- 64.Wuebbens MM, Rajagopalan KV. 1995. Investigation of the early steps of molybdopterin biosynthesis in Escherichia coli through the use of in vivo labeling studies. J Biol Chem 270:1082–1087. doi: 10.1074/jbc.270.3.1082. [DOI] [PubMed] [Google Scholar]
- 65.Leimkühler S, Wuebbens MM, Rajagopalan KV. 2011. The history of the discovery of the molybdenum cofactor and novel aspects of its biosynthesis in bacteria. Coord Chem Rev 255:1129–1144. doi: 10.1016/j.ccr.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Curtis PD, Brun YV. 2014. Identification of essential alphaproteobacterial genes reveals operational variability in conserved developmental and cell cycle systems. Mol Microbiol 93:713–735. doi: 10.1111/mmi.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Workun GJ, Moquin K, Rothery RA, Weiner JH. 2008. Evolutionary persistence of the molybdopyranopterin-containing sulfite oxidase protein fold. Microbiol Mol Biol Rev 72:228–248. doi: 10.1128/MMBR.00041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanjee U, Ogata K, Houry WA. 2012. Direct binding targets of the stringent response alarmone (p)ppGpp. Mol Microbiol 85:1029–1043. doi: 10.1111/j.1365-2958.2012.08177.x. [DOI] [PubMed] [Google Scholar]
- 69.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 70.Tuckerman JR, Gonzalez G, Sousa EH, Wan X, Saito JA, Alam M, Gilles-Gonzalez MA. 2009. An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry 48:9764–9774. doi: 10.1021/bi901409g. [DOI] [PubMed] [Google Scholar]
- 71.Chang AL, Tuckerman JR, Gonzalez G, Mayer R, Weinhouse H, Volman G, Amikam D, Benziman M, Gilles-Gonzalez MA. 2001. Phosphodiesterase A1, a regulator of cellulose synthesis in Acetobacter xylinum, is a heme-based sensor. Biochemistry 40:3420–3426. doi: 10.1021/bi0100236. [DOI] [PubMed] [Google Scholar]
- 72.Qi Y, Rao F, Luo Z, Liang ZX. 2009. A flavin cofactor-binding PAS domain regulates c-di-GMP synthesis in AxDGC2 from Acetobacter xylinum. Biochemistry 48:10275–10285. doi: 10.1021/bi901121w. [DOI] [PubMed] [Google Scholar]
- 73.Weiss G, Fuchs D, Hausen A, Reibnegger G, Werner ER, Werner-Felmayer G, Semenitz E, Dierich MP, Wachter H. 1993. Neopterin modulates toxicity mediated by reactive oxygen and chloride species. FEBS Lett 321:89–92. doi: 10.1016/0014-5793(93)80627-7. [DOI] [PubMed] [Google Scholar]
- 74.Murr C, Widner B, Wirleitner B, Fuchs D. 2002. Neopterin as a marker for immune system activation. Curr Drug Metab 3:175–187. doi: 10.2174/1389200024605082. [DOI] [PubMed] [Google Scholar]
- 75.Hoffmann G, Schobersberger W, Frede S, Pelzer L, Fandrey J, Wachter H, Fuchs D, Grote J. 1996. Neopterin activates transcription factor nuclear factor-kappa B in vascular smooth muscle cells. FEBS Lett 391:181–184. doi: 10.1016/0014-5793(96)00729-6. [DOI] [PubMed] [Google Scholar]
- 76.Gostner JM, Becker K, Fuchs D, Sucher R. 2013. Redox regulation of the immune response. Redox Rep 18:88–94. doi: 10.1179/1351000213Y.0000000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stewart PS, Franklin MJ. 2008. Physiological heterogeneity in biofilms. Nat Rev Microbiol 6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- 78.Heckel BC, Tomlinson AD, Morton ER, Choi JH, Fuqua C. 2014. Agrobacterium tumefaciens ExoR controls acid response genes and impacts exopolysaccharide synthesis, horizontal gene transfer, and virulence gene expression. J Bacteriol 196:3221–3233. doi: 10.1128/JB.01751-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Allen KD, Xu H, White RH. 2014. Identification of a unique radical S-adenosylmethionine methylase likely involved in methanopterin biosynthesis in Methanocaldococcus jannaschii. J Bacteriol 196:3315–3323. doi: 10.1128/JB.01903-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DcpA domain alignments. Alignments performed using ClustalX multiple alignment mode and neighbor-joining (NJ) clustering algorithm. (A) Alignment of the DGC domain of DcpA to characterized DGC proteins. Residues important for catalytic function are outlined in red. (B) Alignment of the DcpA EAL domain to characterized proteins containing EAL domains. Residues important for catalytic function are outlined in red. Download
Additional ΔdcpA phenotypes. (A) Congo red phenotypes of the indicated A. tumefaciens strains. Exponential-phase cultures were normalized to an OD600 of 0.5, and 5 µl of the corresponding culture was spotted onto ATGN minimal medium plates containing 100 µg/ml Congo red and 400 µM IPTG. Pictures were taken after 48 h of growth at 28°C. DGC−, DcpA variant containing catalytic site GGDEF→GGDAF mutation; PDE−, DcpA variant containing catalytic site EAL→AAL mutation. (B) Adherent bacterial biomass grown for 48 h on PVC coverslips quantified by the amount of solubilized crystal violet (CV) at A600, as described in the legend to Fig. 1C. In parallel, the OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the A600/OD600 ratio. The indicated amounts of IPTG were added. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
Intracellular c-di-GMP levels. (A) Quantification of intracellular levels of c-di-GMP in the indicated A. tumefaciens strains. Strains were grown to stationary phase, nucleotides were extracted, and measurements of c-di-GMP were obtained by LC-MS/MS as described in Materials and Methods. All strains were constructed in an extracellular polymeric substance (EPS)-negative strain background (45) in which the genes coding for the polysaccharides succinoglycan, cellulose, cyclic β-1-2 glucans, and curdlan (β-1-3-glucans) have been deleted. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
PruA orthologs complement ΔpruA phenotypes. (A) A. tumefaciens pruA aligned to orthologs from other Rhizobiales and alphaproteobacterial species. Orthologs from C. crescentus CC0417, R. pomeroyi Spo3638, S. meliloti SMc00603, Brucella melitensis bv. abortus BAB1_0721, and E. coli folM were used. Residues important for catalytic function are outlined in red. Alignments were performed using ClustalX multiple alignment mode and NJ clustering algorithm. (B) Adherent bacterial biomass grown for 48 h on PVC coverslips was quantified by the amount of solubilized crystal violet (CV) at A600 as described in the legend to Fig. 1C. In parallel, OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the A600/OD600 ratio. A mutant with no plasmid inserted is indicated (hyphen). Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. (C) Congo red phenotypes of the indicated A. tumefaciens strains. Cultures were normalized to an OD600 of 0.5, and 5 µl of corresponding culture was spotted onto ATGN minimal medium plates containing 100 µg/ml Congo red and 400 µM IPTG. The images were taken after 48 h of growth at 28°C. (D) Predicted PruA protein structure. Structure predicted using PHYRE database (http://www.sbg.bio.ic.ac.uk/phyre2). Leishmania pteridine reductase structure (PDB identifier [ID] 2XOX) is shown for comparison. Alpha helices and beta sheets are highlighted in green and red, respectively. The PruA catalytic tyrosine residue is indicated by a white arrow. Download
PruA enzymatic function necessary for regulation of DcpA DGC/PDE activity. Adherent bacterial biomass grown for 48 h on PVC coverslips quantified by the amount of solubilized crystal violet (CV) at A600 as described in the legend to Fig. 1C. In parallel, the OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the A600/OD600 ratio. IPTG (400 µM) was added to each strain. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
PruR sequence and role in attachment control. (A) Several PruR homologs are missing the cysteine required for molybdopterin binding. A. tumefaciens pruR homologs R. pomeroyi Spo0162, R. capsulatus RCAP rcc01507, R. sphaeroides RSP_0086 were aligned to yedY, a known molybdopterin-binding protein from E. coli. The canonical cysteine is highlighted in red. OR, oxidoreductase; SO, sulfite oxidase. (B) PruA and DcpA are both required for effective PruR complementation. Adherent bacterial biomass grown for 48 h on PVC coverslips quantified by the amount of solubilized crystal violet (CV) at A600 as described in the legend to Fig. 1C. In parallel, the OD600 of nonattached culture was determined to quantify planktonic culture growth, and absorbance was normalized to culture growth by calculating the ratio A600/OD600 ratio. IPTG (400 µM) was added to each strain. Values are results of three independent biological replicates consisting of three technical replicates each. The error bars show 1 SD. Download
2′-O-methyl-5,6,7,8-tetrahydromonapterin biosynthesis and structure. (A) PruA, which does not possess a predicted methyltransferase capability, reduces 7,8-dihydromonapterin to 5,6,7,8-tetrahydromonapterin, which is then methylated by an unknown methyltransferase to generate 2′-O-methyl-5,6,7,8-tetrahydromonapterin. (B) Major fragment ions derived from chemically synthesized 2′-methoxymonapterin during MS/MS analysis. Collisionally induced fragments arising from 2′-O-methylmonapterin observed in both the pterin isolated from A. tumefaciens and the chemically synthesized standard. The 191 m/z fragment was not observed upon analysis of chemically synthesized 3′-O-methylmonapterin (data not shown), providing evidence that the methylation is at the 2′ position in the new pterin from A. tumefaciens. (C) 2′-O-methylmonapterin mass spectra. MS/MS spectrum of the unknown pterin from wild-type A. tumefaciens cells (top) and of chemically synthesized 2′-O-methylmonapterin (bottom). Download
Strains and plasmids.
Oligonucleotides.
Supplemental Materials and Methods. Download