

Abstract
Previous gene-centric analysis of a cow rumen metagenome revealed the first potentially cellulolytic polysaccharide utilization locus, of which the main catalytic enzyme (AC2aCel5A) was identified as a glycoside hydrolase (GH) family 5 endo-cellulase. Here we present the 1.8 Å three-dimensional structure of AC2aCel5A, and characterization of its enzymatic activities. The enzyme possesses the archetypical (β/α)8-barrel found throughout the GH5 family, and contains the two strictly conserved catalytic glutamates located at the C-terminal ends of β-strands 4 and 7. The enzyme is active on insoluble cellulose and acts exclusively on linear β-(1,4)-linked glucans. Co-crystallization of a catalytically inactive mutant with substrate yielded a 2.4 Å structure showing cellotriose bound in the −3 to −1 subsites. Additional electron density was observed between Trp178 and Trp254, two residues that form a hydrophobic “clamp”, potentially interacting with sugars at the +1 and +2 subsites. The enzyme’s active-site cleft was narrower compared to the closest structural relatives, which in contrast to AC2aCel5A, are also active on xylans, mannans and/or xyloglucans. Interestingly, the structure and function of this enzyme seem adapted to less-substituted substrates such as cellulose, presumably due to the insufficient space to accommodate the side-chains of branched glucans in the active-site cleft.
Cellulose from lignocellulosic biomass holds great potential as a future energy source, given its energy-rich composition and abundance on earth. However, accessing the β-(1,4)-linked glucose units of the cellulose polymer is complicated by the recalcitrant nature of its crystalline molecular structure, which still poses a major obstacle in the efficient industrial production of biofuels from plant biomass1,2. Overcoming these issues relies on obtaining a complete understanding of how this conversion efficiently occurs in nature, as well as increasing our knowledge and arsenal of cellulolytic enzymes and microbial cellulolytic systems.
The current perception of microbial cellulose-degradation involves two major accepted paradigms, namely secreted endo- and exocellulase enzymes in aerobic micro-organisms, or dockerin-bound endo- and exocellulases arranged in cellulosomes in anaerobes3. Recently, other scenarios have been identified. These include the discovery of free enzymes in anaerobic, cellulosome-containing systems4,5 as well as an apparently intermediate strategy employed by the thermophillic bacterium Caldicellulosiruptor bescii, which produces a highly effective cellulase composed of several catalytic domains with complementary functions6. Another potentially novel mechanism concerns the presence of a cellulose-targeting Polysaccharide Utilization Locus (PUL) identified in an uncultured Bacteroidetes phylotype (AC2a) from the cow rumen7. A PUL-based mechanism8,9,10 relies on cellulose binding proteins and cellulases which are tethered to the outer membrane of the bacterium. Since the AC2a PUL is the first cellulose-targeting PUL described7, further characterization of its cellulases and their activity on crystalline cellulose is of interest. We have studied one of these cellulases, a member of the glycoside hydrolase family 5 (GH5), referred to as AC2aCel5A.
Enzymes classified as GH5 in the Carbohydrate Active Enzymes database (CAZy) are functionally diverse with 20 experimentally verified activities on various polysaccharides, including xyloglucans, mannans, mixed-linkage β-glucans and cellulose (β-1,4 linked glucose)11. GH5s can be further divided into subfamilies based on their sequence similarity and enzyme specificities12. AC2aCel5A is affiliated with subfamily 4, a polyspecific family which typically includes extracellular bacterial enzymes that exhibit one or more activities categorized as endoglucanase, xyloglucan-specific endoglucanase, xylanase, and licheninase. GH5 enzymes share a common (β/α)8-barrel fold, which contains two conserved catalytic glutamic acid residues at the C-terminal ends of the fourth and seventh β-strands. As endo-cellulases are important constituents in commercial cellulase mixtures, the discovery and characterization of additional cellulolytic GH5 enzymes from previously non-accessible biodiversity may prove fruitful for the development of better biomass conversion technology.
Here, we present the three dimensional structure and biochemical characterization of AC2aCel5A. The PUL-derived GH5 cellulase was discovered in the genome of an uncultured Bacteroidetes phylotype reconstructed from a cow rumen metagenome, which was previously assigned to the order Bacteroidales by genome-wide alignment against the NCBI13,14. The gene was synthesized for expression in Escherichia coli with a C-terminal His6-tag for ease of purification. The native structure was solved to 1.8 Å resolution and revealed a narrow substrate-cleft, likely determining the specificity of the enzyme for linear and less substituted substrates.
Results
Structural features of AC2aCel5A
Wild-type AC2aCel5A contains a predicted N-terminal signal peptide, which was removed to simplify over-expression of the 405-residue recombinant protein, including mature AC2aCel5A (397 residues) followed by an eight residue C-terminal His6-tag. The structure of AC2aCel5A was solved to 1.8 Å, and contained two molecules in the asymmetric unit. The final model comprised residues 9–397; no electron density was observed for the nine N-terminal residues nor for the final eight C-terminal residues, including the His6-tag. The data collection and refinement parameters are summarized in Table 1. The enzyme displays the typical (β/α)8 barrel structure associated with the GH5 family (Fig. 1a). This canonical (β/α)8 fold is preceded by an extra alpha-helix (Fig. 1a), as was also observed in its close structural homologues, cellulase EngD from Clostridium cellulovorans (PDB id 3NDY)15 and CelAcd from Piromyces rhizinflata (PDB id 3AYR)16. Additionally, the structure includes six short helices, one in the N-terminus and five in the loop regions between the canonical strands and helices (Fig. 1a; α1′–α6′).
Table 1. Crystal data, data-collection statistics and refinement data.
| Dataset | AC2aGH5 native structure | AC2aGH5_E172A cellotriose complex |
|---|---|---|
| PDB Code | 4YHE | 4YHG |
| Data Collection | ||
| Beamline | ID29 (ESRF) | ID29 (ESRF) |
| Wavelength (λ) | 0.9724 | 0.9791 |
| Space Group | P 21 21 21 | P 21 21 21 |
| Unit-cell parameters (Å, °) | a = 85.5, b = 100.7, c = 101.9 | a = 84.3, b = 99.6, c = 102.5 |
| α = β = γ = 90 | α = β = γ = 90 | |
| Resolution (Å) | 65.5 - 1.85 (1.9 - 1.85) | 54.5-7.6 (2.5-2.40) |
| Unique reflections | 74 499 (4027) | 34 325 (4927) |
| Multiplicity | 3.7 (2.9) | 5.2 (5.1) |
| Completeness (%) | 98.6 (90.8) | 99.8 (99.8) |
| Mean I/σI | 8.9 (2.9) | 10.2 (3.1) |
| Rmeasa | 0.094 (0.36) | 0.116 (0.551) |
| Refinement statistics | ||
| Rcryst/Rfree (%)b | 17.8 (21.2) | 18.5 (22.8) |
| R.m.s.d. bonds (Å) | 0.011 | 0.010 |
| R.m.s.d. angles (°) | 1.4 | 1.3 |
| B-factor | ||
| protein/solvent/ligands (Å2) | 25.0 / 35.5 | 42.3 / 35.0 / 55.2 |
| Number of atoms | ||
| Protein | 6155 | 5941 |
| Solvent | 955 | 232 |
| Cellotriose | 34 | |
| Ramachandran plot (%)c | ||
| Favorable regions | 97.8 | 97.2 |
| Additionally allowed regions | 1.9 | 2.5 |
| Outliers | 0.3 | 0.3 |
Values in parentheses are for the highest resolution shells
aRmeas defined by Diederichs & Karplus42.
bRcryst = Σhkl ||Fo| - |Fc|| / Σhkl|| Fo | where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree is calculated from a randomly chosen 5% set of all unique reflections not used in refinement.
cDefined using MolProbity43.
Figure 1. Overall structure and features of AC2aCel5A.

a) Cartoon representation, rainbow colored blue to red from residue 9 to residue 397. Canonical secondary structure elements are labeled as α1–α8 for alpha-helices, and β1–β8 for beta-sheets. The conserved additional N- terminal helix is labeled α0, and the six remaining helices additional to the canonical fold are labeled α1′–α6′. b) Overall structure highlighting the side-chains of strictly conserved residues in the catalytic centers of family 5 GHs, including the catalytic acid, Glu172 and the catalytic nucleophile/base, Glu30315,41. c,d) Surface-views highlighting the active-site groove. The catalytic glutamates are colored red, and other strictly conserved residues highlighted in panel b are colored blue.
The six closest structural homologues, which include all the biochemically characterized members of subfamily GH5_4, were identified using the Dali server17 and were used to produce a structure-based multiple sequence alignment using the PROMALS3D server18 (Supplementary Fig. S1). This allowed identification of the conserved catalytic residues, i.e. Glu172 and Glu303, acting as the catalytic acid and nucleophile/base, respectively. Six other strictly conserved residues in the catalytic centers of GH5s15 were also identified (Fig. 1b). A surface projection shows the narrow groove stretching across the enzyme, with the active-site located in its center (Fig. 1c–d). The roles of Glu172 and Glu303 as catalytic residues were confirmed by site-directed mutagenesis experiments, showing that AC2aCel5A_EE172A and AC2aCel5A_EE303A were both inactive in the standard carboxymethylcellulose (CMC) assay.
Cellotriose complex
Co-crystallization of the E172A mutant with cellotetraose resulted in the complex structure solved to 2.4 Å resolution, with two molecules in the asymmetric unit. Additional electron density was observed in the active-site cleft of molecule A, and was refined as a cellotriose molecule bound to the −3 to −1 subsites (Fig. 2a–b). No electron-density for a bound ligand could be observed in the B molecule, which also lacked electron density for several main-chain residues, namely 34–45 and 112–127. These residues correspond to the loop regions between β1 and α1, and between β3 and α2’, respectively, and have been omitted from the final model. Even though the ligand density appears to fit a cellotriose molecule, it is likely that a cellotetraose molecule is present in the complex as the enzyme only produces dimers from cellotetraose7. The observed cellotriose density could reflect three glucose moieties of a tetramer, where the fourth moiety could be positioned in the putative −4 position and be disordered. This is supported by the observation that the electron density of the -3 bound moiety is incomplete towards O4 (Fig. 2b).
Figure 2. Structure of AC2aCel5A in complex with cellotriose.

a) Surface view showing cellotriose (yellow carbons) in the active site-cleft with its 2Fo − Fc map contoured at 1.0 σ. The aromatic residues Trp178 and Trp254 are indicated in red to demonstrate their positions in the cleft. b) Close-up view of the bound cellotriose with its 2Fo − Fc map contoured at 1.0 σ, indicating the −3 to −1 subsites c) Divergent (wall-eyed) stereo view detailing un-modelled 2Fo − Fc electron density in the cleft contoured at 1.0 σ. The side chains of Trp178 and Trp254 are shown as sticks and colored red. d) Divergent (wall-eyed) stereo view of ligand-protein interactions determined using LigPlot+. Green residues hydrogen-bond to cellotriose (yellow), whereas magenta residues interact with the ligand by hydrophobic and Van der Waals forces. The position of the side chain of Glu172 (cyan) was determined by superposition of the wild-type structure.
Furthermore, in both the apo- and the complex structure, extra electron density was also observed close to, and between Trp178 and Trp254 in the substrate-binding cleft of both chain A and B (Fig. 2c). The electron density was insufficient to allow meaningful interpretation, but did not appear to resemble any of the components found in the cryoprotectant or mother liquor. In GH5 complexes where either a tetramer or pentamer ligand is bound across the catalytic site, a tryptophan corresponding to Trp178 of AC2aCel5A is always interacting with the sugar moiety bound in the +1 subsite and in some cases also the sugar in the +2 subsite (PDB ids; 4HU0, 3AZT19, 2CKR, 1H5V, 1ECE20, 3QHN21 and 4OOZ22). The aromatic residue corresponding to Trp254 in AC2aCel5A is much less conserved; this residue’s role is most commonly appears to be taken by tyrosine (2CKR, 1ECE, 3QHN), whereas also proline (4HU0), phenylalanine (3AZT) and histidine (4OOZ) occur in a similar position of the active-site cleft. 1H5V lacks a residue corresponding to Trp254. While the role of the Trp178-Trp254 “clamp” in ligand binding seems obvious, it seems unlikely that the density, observed in both the apo- and the ligand structure, reflects substrate. It is worth noting that the C-terminus of an adjacent B molecule is located in close proximity to the active-site cleft of molecule A, and vice versa. It is therefore likely that the electron density in the Trp-Trp “clamp”, as well as the additional density seen outside the clamp, reflects a non-specific interaction of the C-terminal His6–tag which protrudes towards the active site cleft.
LigPlot+23 was used to investigate the interactions of the enzyme with the cellotriose molecule, and the estimated interactions are shown in Fig. 2d. The glucose moiety bound in the −1 subsite is stabilized by hydrogen bonding from its O2 group to the catalytic Glu303 residue, as well as the strictly conserved Asn171. O3 interacts with His111 as well as Asn30, Asp33 and His112 via a water molecule, HOH25. The same water also interacts with O5 of the glucose bound in the -2 subsite. The O2 of the latter sugar has hydrogen-bond type interactions with Trp339, Asn341 and Asp349; Asp349 also interacts with the O3 together with Asn30. Unlike the sugars bound in the −2 and −1 subsites, the sugar bound in the −3 subsite involves no hydrogen bonds between the ligand and the protein. Its binding seems to be mediated by stacking interactions with Trp45. Other hydrophobic and van der Waals contacts contributing to ligand binding involve the strictly conserved His249 and Tyr251, which interact with the −1 glucose moiety, and Phe115, interacting with the −2 sugar. Superposition of the native structure on the cellotriose complex placed a carboxyl oxygen of the catalytic acid, Glu172, at 1.7 Å of O1 of the glucose bound in the −1 subsite.
Biochemical characterization
As previously reported7, AC2aCel5A is an endoglucanase active on β-(1,4) glycosidic bonds between glucose units in soluble and insoluble cellulosic substrates and in linear β-glucans. Further characterization and quantification of enzyme activities revealed high activities on linear β-(1,4) glucans, whereas the enzyme displayed only trace activity on soluble tamarind xyloglucan and no activity could be detected on solubilized Birchwood xylan (Table 2). Using the standard CMC assay, the optimum temperature of the enzyme was found to be 40 °C, correlating with the 38–40 °C environment of the cow rumen24 (Fig. 3). However, the enzyme showed activity over a relatively broad temperature range, retaining close to 60% of maximum activity between 20 °C and 50 °C. Assays with different buffer systems of various pH values also showed a broad activity range. Maximum activity was observed at pH 5.0, while over 60% of maximum activity was maintained between pH 4.5 and 8.0.
Table 2. Structural comparison of AC2aCel5A with its six closest structural homologues identified using the DALI server17, along with reported enzyme activities.
| Structural comparison | Enzymatic activities | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dali Z-score | RMSD (Å) | % Id | CMC (U/mg) | β-glucan (U/mg) | Filter paper (U/mg) | Avicel (U/mg) | Lichenan (U/mg) | Xylan (U/mg) | Xyloglucan (U/mg) | Reference | |
| Bacteroidetes AC2a Cellulase Cel5A | − | − | − | 216.8 | 1471.4 | 0.152a | 0.115a | 839.9 | nd | trace | This study and 7 |
| Paenibacillus pabuli Xyloglucanase XG5 (2JEP) | 49.2 | 1.6 | 34 | nd | nd | − | nd | nd | nd | 8700 | 33 |
| Clostridium cellulovorans Cellulase EngD (3NDY) | 47.0 | 1.7 | 33 | 15 | 42 | − | 0.017 | − | 0.5 | 36 | 15 |
| Bacteroides ovatus Xyloglucanase BoGH5A (3ZMR) | 46.6 | 1.9 | 32 | nd | nd | − | − | nd | − | 514.2 | 44 |
| Clostridium thermocellumCellulase Lic26A-5E (4IM4) | 46.0 | 1.6 | 32 | Active | 1200 | − | − | − | Active | − | To be published, NZYTechb |
| Clostridium cellulolyticum Cellulase CelCCA (1EDG) | 45.9 | 1.9 | 32 | 101.3 | 104.3 | − | 0.028 (0.124)d | 79.2 | 10.1 | − | 45,c |
| Piromyces rhizinflata Cellulase CelAcd (3AYR) | 43.2 | 1.8 | 30 | 344.9 | 576 | 0.64 | 1.39 | 542.5 | 106.2 | − | 47 |
RMSD; root-mean-square deviation of C-alpha atoms. The structural homologues are sorted based on the Z-score obtained in the DALI search. One Unit of enzyme activity was defined as the amount of enzyme releasing 1 μmol of reducing sugar equivalents per minute. “nd” means not detected, whereas a hyphen, “-”, indicates “not tested”.
aμmol reducing sugar equivalents calculated as μmol cellotriose + μmol cellobiose + μmol glucose, quantified by HPAEC-PAD7.
bGH5 domain from CtLic26A-Cel5E. Described as multi-functional cellulase, mannanase and xylanase by Bianchetti et al. in the PDB entry file; β-glucan activity for the Cel5E domain reported by NZYTech (1649-038 Lisboa, Portugal/www.nzytech.com).
cU/mg calculated from U/μmol reported in45.
dFor this enzyme data for both the full length protein (in parentheses), and the catalytic domain only were published.
Figure 3. Biochemical characterization of AC2aCel5A.
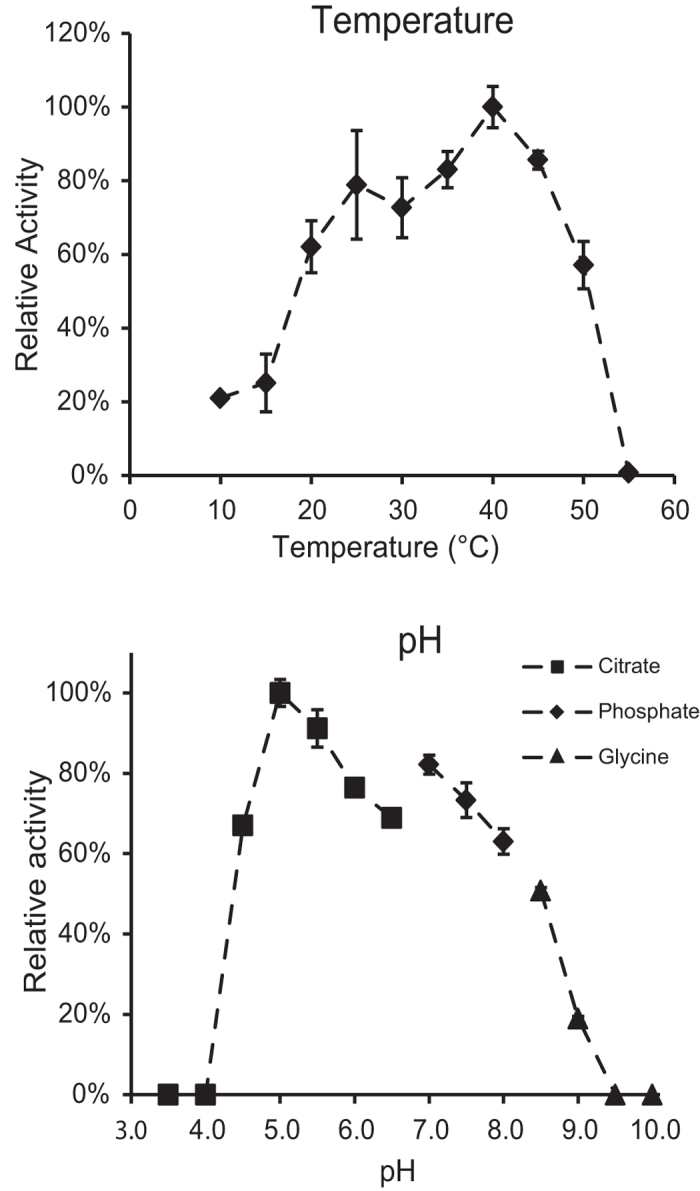
Enzyme activity is reported as the amount of reducing sugars released in the standard CMC assay, relative to optimal temperature (top panel) or pH (bottom panel) conditions. Error bars represent standard deviations between three replicates.
Comparison to structural homologues
The six closest structural homologues of AC2aCel5A previously identified with the Dali server were subjected to further investigation (Table 2). The selected structures had root-mean-squared deviations (R.m.s.d.) of less than 1.9 Å when compared with AC2aCel5A. Two of the homologues have been characterized as xyloglucanases (PDB ids: 2JEP and 3ZMR), whereas the four others are identified as cellulases (PDB ids: 3NDY, 4IM4, 1EDG and 3AYR) (Table 2). The xyloglucanase homologues are reported as strictly specific for xyloglucan, whereas the cellulase-type homologues generally seem more promiscuous in their substrate specificities. For example, EngD (PDB id 3NDY) has comparable activity on xyloglucan (1,6-xylose substituted β-1,4 glucan), CMC and Barley β-glucan, and also showed activity on birchwood xylan (β -1,4 xylose substituted with 10.2% hexuronic acids25). CelCCa (PDB id 1EDG) from Clostridium thermocellum and CelAcd (PDB id 3AYR) from Piromyces rhizinflata were not tested on xyloglucan, but showed activities on insoluble xylan from Sigma, and soluble oat spelt xylan (substituted with arabinose (9%), glucose (7%), galactose (1%)25), respectively. Conversely, AC2aCel5A is specific for unsubstituted β-1,4 glucans and CMC (cellulose substituted with relatively small carboxymethyl groups), with only trace activity on xyloglucan and no detectable activity on birchwood xylan.
A comparison of the active-site clefts of the GH5s shows that AC2aCel5A has a narrower active site cleft than its homologues, especially in the vicinity of the −3 to −1 sub-sites (Fig. 4). This is primarily caused by the loop region between β3 and α3’ (red surface), which protrudes into the substrate-binding cleft and which is extended in AC2aCel5A, compared to the examined homologues (Supplementary Fig. S1, residues 115–126). The figure also indicates the aromatic residues, corresponding to the putative +1 site of AC2aCel5A, that are structurally conserved in these close homologues (yellow surface). Fig. 5a shows a detailed view of the differences between the surface projections of AC2aCel5A (black mesh) and the cellulase CelAcd (blue), highlighting that this homologue has a wider active site cleft. The picture further shows that the active site cleft of AC2aCel5A is far too narrow to accommodate a xyloglucan tetramer due to the extended loop between β3 and α3’ (red backbone and mesh), whereas CelAcd has enough space to accommodate this branched oligosaccharide. Fig. 5b shows that a true xyloglucanase such as XG5 from Paenibacillus pabuli also has a wider cleft than AC2aCel5A but that this cleft is narrower compared to the promiscuous CelAcd. In the XG5 xyloglucanase, the cleft seems more optimally shaped to harbour the xyloglucan fragment, providing many tight interactions.
Figure 4. Active site grooves in related GH5 endoglucanases.

Catalytic centers are indicated by the two catalytic glutamates whose surfaces are colored red. The surface of the residues lining the +1 and/or +2 subsites, analogous to Trp178 and Trp254 in AC2aCel5A are colored yellow (see text for details). The 115-126 loop in AC2aCel5A is coloured red. The −3 to −1 subsites are indicated by the superimposed cellotriose molecule from AC2aCel5A.
Figure 5. Structural superpositions.

a) CelAcd from P. rhizinflata (PDB id 3AYS) (blue) structurally aligned by PyMOL to AC2aCel5A (black mesh). The main chain of the loop region of AC2aCel5A between β3 and α2’ is shown and colored red. The ligands shown are cellotriose (yellow) binding to the −3 to −1 site of AC2aCel5A and a xyloglucan tetramer (green) which was positioned by structural superposition of the ligand-containing structure of P. pabuli XG5 xyloglucanase (PDB id 2JEQ). The xyloglucan ligand is an XXLG tetramer, where G, X and L denote unsubstituted D-Glcp, α-D-Xylp-(1,6)-D-Glcp, and β-D-Galp-(1,2)-α-D-Xylp(1,6)-D-Glcp, respectively. b) Shows the same structural alignment as in panel a), with the same elements and coloring, but with the P. pabuli XG5 xyloglucanase, shown in orange, instead of CelAcd. The xyloglucan tetramer binds to subsites −1 to −4.
Discussion
AC2aCel5A is an endo-acting cellulase that is encoded within the first reported cellulolytic PUL, originating from an uncultured Bacteroidetes phylotype. Biochemical data presented in this study demonstrate that optimal conditions for AC2aCel5A activity correspond well with the cow rumen environment. AC2aCel5A was evolutionarily distinct compared to its nearest structural homologues (30–34% sequence identity, Table 2), whereas its nearest sequence homologue was a putative GH5 recovered from a goat rumen metagenome (51% sequence identity, accession number: AIF26005). According to the CAZy subfamily classification proposed by Aspeborg et al.12, AC2aCel5A and all of the examined homologues (Table 2) belong to subfamily GH5_4. Among these subfamily GH5_4 enzymes, the deeply branched AC2aCel5A is dissimilar in that it lacks activity on both xylan and xyloglucan. The interactions between cellotriose and the -3 to -1 subsites of AC2aCel5A are similar to the interactions observed in enzyme-substrate complexes of EngD15 (PDB id 3NDZ) and CelAcd16 (PDB id 3AYS). Nonetheless, AC2aCel5A is unique in that it has an exceptionally narrow active site cleft, due to an extended loop region that seems well adapted to (only) acting on less-substituted β-glucans.
AC2aCel5A contains a tryptophan “clamp” in its putative +1 and +2 subsites, analogous to what has been observed in other glycoside hydrolases acting on insoluble polysaccharides26,27. This aromatic pair is not conserved in all GH5s (see text above), but all of the examined structural homologues except the Bacteroides ovatus xyloglucanase, BoGH5A (PDB id 3ZMR), do contain two structurally conserved aromatic residues that form a similar substrate-binding “clamp” (Fig. 4). Although the complex structure of AC2aCel5A does not provide direct new insight into the interaction between the two aromatic residues and the substrate, the presence of electron density in the “clamp” in both the apo and complex structures suggests a potential for strong stacking interactions, and likely indicates the enzyme’s putative +1 (and +2) subsites.
This study presents new insights into the biology of an as-yet uncultured Bacteroidetes-affiliated phylotype (AC2a). Collectively, the AC2aCel5A enzyme structure and activity data suggest specificity towards linear and less substituted β-(1,4)-linked glucans, and strengthens previous hypotheses that the PUL encoding AC2aCel5A is cellulolytic. Ongoing studies directed towards elucidating the structures and functions of other proteins encoded by this PUL (e.g. SusD and SusE cellulose-binding proteins) will enhance our understanding of its polysaccharide degradation potential.
Methods
Protein discovery and expression
The GH5 enzyme (AC2aCel5A) was identified and produced as previously described7. In brief, the gene was synthesized without its predicted signal peptide (residues 1–18), cloned into the pNIC-CH vector28 and over-expressed in Escherichia coli. The C-terminally His6-tagged protein was purified to near homogeneity by immobilized metal affinity chromatography, and the purity was assessed by SDS-PAGE. The protein concentration was estimated by A280 using the molar extinction coefficient calculated from the protein sequence. The protein was stable at concentrations around 20 mg/ml in 20 mM Tris-HCl pH 8 with 0.2 M NaCl, at 4 °C, for several months.
Site-directed mutagenesis
The QuickChange II site-directed mutagenesis kit (Agilent) was used to mutate the catalytic glutamic acid residues (E172 and E303) to alanines, using the following primers; E172AF; GGT TTT TGA AAC CCT GAA TGC AAT TCA GGA TGG TGA TTG GG, E172AR; CCC AAT CAC CAT CCT GAA TTG CAT TCA GGG TTT CAA AAA CC, E303AF; GCC GGT TTA TGG TGC ATT TGG TGC CGT TCG, E303AR; ACG AAC GGC ACC AAA TGC ACC AAA ATA AAC CGG.
Crystallization, diffraction data collection and structure determination
Native crystals were produced by screening sitting drop vapor diffusion conditions using the JCSG+ 1 kit (Molecular Dimensions; Altamonte Springs, FL, USA), with a reservoir volume of 100 μL, and a drop comprising 0.5 μL well solution +0.5 μl purified protein solution (10.2 mg/ml in 20 mM Tris-HCl pH 8.0, 0.2 M NaCl). Crystals grew overnight in several conditions, with highest quality crystals forming in conditions consisting of 0.1 M sodium cacodylate pH 6.5, 40% v/v 2-Methyl-2,4-pentanediol and 5% w/v PEG8000. Crystals for the ligand bound complex were obtained by co-crystallization of the catalytically inactive AC2aCel5A E172A mutant in conditions consisting of 0.2 M potassium nitrate and 20% w/v PEG 3350. The reservoir volume was 200 μL and the 2 μL drop contained a 1:1 mixture of well solution and protein solution (11 mg/ml protein in 20 mM Tris-HCl pH 8.0, 0.2 M NaCl, 5 mM cellotetraose). Crystals were mounted in loops and flash-frozen with liquid nitrogen. X-ray diffraction data was collected at the ID29 beamline at the European Synchrotron Radiation Facility (ESRF), to a resolution of 1.8 Å for the apo protein, and 2.1 Å for the complex. The apo data was processed using iMOSFLM29, Aimless30 and tools in the CCP4i package31. The structure was solved by molecular replacement with Phaser32 using a poly-alanine model of the structure of a GH5 family xyloglucanase from Paenibacillus pabuli33 (PDB-entry 2JEP). The initial model was built using the autobuild function of PHENIX34 and further refined using PHENIX, RefMac535,36 and manual rebuilding in Coot37. The dataset for the complex was processed with the XDS package38 and scaled using Scala39, cutting the data to 2.4 Å. The structure was solved by molecular replacement using the apo structure of AC2aCel5A with MolRep, and the structure was refined using RefMac5 and manual rebuilding in Coot. All protein structure figures were produced in PyMOL.
Enzyme characterization
The substrate specificity of AC2aCel5A had previously been determined using Azurine-Crosslinked Polysaccharide (AZCL) substrates, along with activity on insoluble cellulose substrates7. In this study, further characterization of the enzymatic activity was performed using the soluble β-(1,4) linked glucan substrates carboxymethylcelullose (CMC) (Sigma-Aldrich), barley β-glucan (Megazyme), tamarind xyloglucan (Megazyme), lichenan (Sigma-Aldrich), and Birchwood Xylan (Carl Roth). The standard reaction using CMC contained 20 mM BisTris buffer pH 6.5, 25 nM enzyme, 20 mM CaCl2, and 10 mg/ml substrate in a total volume of 200 μl. Enzyme was added to pre-heated assay mixtures and the reactions were incubated at 40 °C, with 900 rpm vertical shaking. 100 μl sample was taken after 10 minutes, and added to 100 μL DNS reagent40. The amount of reducing ends released were determined as glucose equivalents using the DNS reducing-end assay and a glucose standard curve. Assays for activity on barley β-glucan, lichenan, xylan and xyloglucan were performed with 0.5% substrate (w/v), and 10, 10, 200 and 200 nM enzyme load, respectively. A Unit of enzyme activity was defined as the amount of enzyme releasing one μmol of glucose equivalents per minute.
Additional Information
How to cite this article: Naas, A.E. et al. Structural Features of a Bacteroidetes-Affiliated Cellulase Linked with a Polysaccharide Utilization Locus. Sci. Rep. 5, 11666; doi: 10.1038/srep11666 (2015).
Supplementary Material
Acknowledgments
We are grateful for support from The Research Council of Norway’s FRIPRO program (214042) and for synchrotron travel support (216625/F50), as well as the European Research Commission Starting Grant Fellowship (awarded to PBP; 336355 - MicroDE). AM was supported by a grant from the Research Council of Norway (190965). This study was also supported by the South-Eastern Norway Regional Health Authority (Grants No. 2012085 and 2015095; Regional Core Facility for Structural Biology to BD). We would also like to thank the European Synchrotron Radiation Facility staff for help and beamtime at beamline ID29 (project MX-1468).
Footnotes
The authors declare no competing financial interests.
Author Contributions P.B.P. and V.G.H.E. proposed this project. P.B.P., V.G.H.E. and A.K.M. designed the experiments and supervised the project. A.E.N. and B.D. did the experiments. A.E.N., B.D. and A.K.M. analyzed the data. A.E.N., A.K.M., V.G.H.E. and P.B.P. contributed to analysis of the results and paper writing.
References
- Horn S. J., Vaaje-Kolstad G., Westereng B. & Eijsink V. G. Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Marcuschamer D., Oleskowicz-Popiel P., Simmons B. a & Blanch H. W. The challenge of enzyme cost in the production of lignocellulosic biofuels. Biotechnol. Bioeng. 109, 1083–1087 (2012). [DOI] [PubMed] [Google Scholar]
- Wilson D. B. Three microbial strategies for plant cell wall degradation. Ann. N. Y. Acad. Sci. 1125, 289–297 (2008). [DOI] [PubMed] [Google Scholar]
- Cai S. et al. Cellulosilyticum ruminicola, a newly described rumen bacterium that possesses redundant Fibrolytic-protein-encoding genes and degrades lignocellulose with multiple carbohydrateborne fibrolytic enzymes. Appl. Environ. Microbiol. 76, 3818–3824 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. et al. Omics-based interpretation of synergism in a soil-derived cellulose-degrading microbial community. Sci. Rep. 4, 1–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunecky R. et al. Revealing Nature’s Cellulase Diversity: The Digestion Mechanism of Caldicellulosiruptor bescii CelA. Science (80-.). 342, 1513–1516 (2013). [DOI] [PubMed] [Google Scholar]
- Naas A. E. et al. Do rumen Bacteroidetes utilize an alternative mechanism for cellulose degradation? MBio 5, e01401–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropatkin N., Martens E., Gordon J. & Smith T. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16, 1105–1115 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope P. B. et al. Metagenomics of the svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization Loci. PLoS One 7, e38571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie a K. et al. Two SusD-like proteins encoded within a polysaccharide utilization locus of an uncultured ruminant Bacteroidetes phylotype bind strongly to cellulose. Appl. Environ. Microbiol. 78, 5935–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspeborg H., Coutinho P. M., Wang Y., Brumer H. & Henrissat B. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evolutionary Biology 12, 186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess M. et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–7 (2011). [DOI] [PubMed] [Google Scholar]
- Pruitt K. D., Tatusova T. & Maglott D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchetti C. M. et al. Structure, dynamics, and specificity of endoglucanase D from Clostridium cellulovorans. J. Mol. Biol. 425, 4267–4285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C. W. et al. Substrate binding of a GH5 endoglucanase from the ruminal fungus Piromyces rhizinflata. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 67, 1189–1194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. & Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J., Kim B. H. & Grishin N. V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.-H. et al. Diverse substrate recognition mechanism revealed by Thermotoga maritima Cel5A structures in complex with cellotetraose, cellobiose and mannotriose. Biochim. Biophys. Acta 1814, 1832–1840 (2011). [DOI] [PubMed] [Google Scholar]
- Sakon J., Adney W. S., Himmel M. E., Thomas S. R. & Karplus P. A. Crystal Structure of Thermostable Family 5 Endocellulase E1 from Acidothermus cellulolyticus in Complex with Cellotetraose †,‡. 2960, 10648–10660 (1996). [DOI] [PubMed]
- Kim H.-W. & Ishikawa K. Functional analysis of hyperthermophilic endocellulase from Pyrococcus horikoshii by crystallographic snapshots. Biochem. J. 437, 223–230 (2011). [DOI] [PubMed] [Google Scholar]
- Kim M.-K. et al. Structure-based investigation into the functional roles of the extended loop and substrate-recognition sites in an endo-β-1,4-D-mannanase from the Antarctic springtail, Cryptopygus antarcticus. Proteins 82, 3217–3223 (2014). [DOI] [PubMed] [Google Scholar]
- Laskowski R. A. & Swindells M. B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011). [DOI] [PubMed] [Google Scholar]
- Weimer P. J., Russell J. B. & Muck R. E. Lessons from the cow: what the ruminant animal can teach us about consolidated bioprocessing of cellulosic biomass. Bioresour. Technol. 100, 5323–31 (2009). [DOI] [PubMed] [Google Scholar]
- Hespell R. & Cotta M. Degradation and utilization by Butyrivibrio fibrisolvens H17c of xylans with different chemical and physical properties. Appl. Environ. Microbiol. 61, 3042–3050 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivula A. et al. Tryptophan 272?: an essential determinant of crystalline cellulose degradation by Trichoderma reesei cellobiohydrolase Cel6A. 429, 341–346 (1998). [DOI] [PubMed]
- Watanabe T. et al. Aromatic residues within the substrate-binding cleft of Bacillus circulans chitinase A1 are essential for hydrolysis of crystalline chitin. Biochem. J. 376, 237–244 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidis C. & de Jong P. J. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye T. G. G., Kontogiannis L., Johnson O., Powell H. R. & Leslie A. G. W. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D. Biol. Crystallogr. 67, 271–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. & Murshudov G. N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster T. M. et al. Characterization and three-dimensional structures of two distinct bacterial xyloglucanases from families GH5 and GH12. J. Biol. Chem. 282, 19177–19189 (2007). [DOI] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N., Vagin A. A. & Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D: Biological Crystallography 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- Murshudov G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G. & Cowtan K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. in Acta Crystallographica Section D: Biological Crystallography 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- Miller G. L. Use of dinitrosalicyclic reagent for determination of reducing sugar. Anal. Chem. 31, 426– 428 (1959). [Google Scholar]
- Davies G. J. et al. Snapshots along an enzymatic reaction coordinate: analysis of a retaining beta-glycoside hydrolase. Biochemistry 37, 11707–11713 (1998). [DOI] [PubMed] [Google Scholar]
- Diederichs K. & Karplus P. A. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269–275 (1997). [DOI] [PubMed] [Google Scholar]
- Davis I. W. et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsbrink J. et al. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierobe H. P. et al. Characterization of endoglucanase A from Clostridium cellulolyticum. J. Bacteriol. 173, 7956–7962 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducros V. et al. Crystal structure of the catalytic domain of a bacterial cellulase belonging to family 5. Structure 3, 939–49 (1995). [DOI] [PubMed] [Google Scholar]
- Liu J.-H., Tsai C.-F., Liu J.-W., Cheng K.-J. & Cheng C.-L. The catalytic domain of a Piromyces rhizinflata cellulase expressed in Escherichia coli was stabilized by the linker peptide of the enzyme. Enzyme Microb. Technol. 28, 582–589 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.