

Abstract
Schwartz–Jampel syndrome is a very rare congenital myotonic syndrome with typical phenotypic and electrophysiological features. Diagnosis is made by awareness into the typical phenotypic characters.
Keywords: Malignant hyperthermia, myotonia, Schwartz–Jampel syndrome
Introduction
This condition also called as chondrodystrophic myotonia was first described in the year 1962 by Schwartz and Jampel as congenital blepharophimosis associated with unique generalized myopathy.[1,2] The prevalence is <1/million and total reported cases are <100.[3] This is inherited as autosomal dominant and autosomal recessive. Most patients become symptomatic between 3 years and 10 years. They present with typical phenotypic features, characterized by short stature, mask-like facies, epicanthic fold, receding chin, upturned nose, long philtrum, short neck, pinched face, low-set ears, high arched palate, prominent muscles, and high pitched voice. The diagnostic findings are typical facial appearance, muscle hypertrophy, and continuous muscle fiber activity.[2] They have slow movements with arms semi-flexed, toe walking in addition to difficulty in opening the mouth, mild kyphosis, contractures at elbow, spine, pelvis, metaphyseal deformities, lumbar lordosis, limited movement in major joints, other complications like hydrocephalus, carpal tunnel syndrome, myelopathy, recurrent infections, stridor and mental retardation etc. But the radiological assessment of bones generally does not yield osseous or epiphyseal changes. However, normal intelligence and normal skeletal system is also known. They show myotonia clinically and electrophysiologically. In general, biochemistry, cytogenetics, and histology are not very helpful. Myopathic, neurogenic, and normal histopathological features are reported. Immunohistochemistry showing abnormal fibers with accumulation of desmin, vimentin, titin with fast, slow, fetal, and embryonic isoforms suggesting regeneration, type 1 fiber predominance and fiber type grouping suggestive of re-innervation can also be seen. Electron microscopy shows abnormalities in the form of dilated T system, Z band streaming, dilatation of mitochondria and also denervation atrophy. Interruption in sarcomere including the Z disc, focal disarray of myofibrils, increased subsarcolemmal sarcoplasm, vacuoles in between bundles of myofibrils, abnormal thin filaments, clusters of glycogen, myelinated materials are also reported. This disease has been mapped to Schwartz–Jampel syndrome locus on chromosome 1(1p34-p36.1).[3] Electrophysiology shows normal motor sensory conductions with characteristic electromyographic features in the form of prolonged and persistent discharges following needle insertion, repetitive discharges, high-frequency discharges, after discharges, dive-bomber sound, and normal motor units. Complex repetitive discharges consisting of potentials which are closely time locked are seen as a result of ephaptic impulse transmission between fibers.[4,5,6] Different patients, however, can show different patterns with a spectrum varying from typical myotonic discharges to discharges with no variation in amplitude and frequency or combination. Bizarre high-frequency patterns are also reported.[6,7] The discharges are from muscle as they cannot be suppressed by curare, but agents that block sodium channels in muscle like procainamide inhibit it like in Isaac's syndrome. The disease is caused by mutation in perlecan, a heparitin sulfate proteoglycan which is in the basement membrane of muscles and cartilage. Loss of its function causes altered clustering of Acetylcholine esterase and abnormal expression of ion channels. Treatment with procainamide or mexiletene can be useful, but no benefit is seen with diphenylhydantoin, diazepam, barbiturates, etc. Early treatment with carbamazepine is reported to have helped few patients.[8] It is important to recognize this condition as they are prone for fatal hyperthermia following anesthesia.[2] Prenatal diagnosis is possible in the mid trimester ultrasound by specifically looking for constant flexion of fingers, decreased fetal activity, and shortening and bowing of femurs. Other myotonic syndromes like congenital myotonic dystrophy, dominant and recessively inherited myotonia congenita and paramyotonia congenita are phenotypically and genotypically distinct. Freeman–Sheldon syndrome is a close mimic with whistling face, puckered lips, microstomia, long philtrum, and dimpled chin. There is ulnar deviation of the hand causing windmill vane configuration. Marden–Walker syndrome is associated with immobile facies, blepharophimosis, mental retardation, congenital joint contractures, and failure to thrive.[2]
Case Report
A 12-year-old male child born to nonconsanguineous parents presented with a history of delayed cry, normal motor development but never learned to run. From the age of 6 years, he was experiencing pain and stiffness of both the legs while walking fast especially over the calf and thighs which improved with rest. But he did not have any buckling or cramps. He also experienced difficulty in climbing stairs in the form of stiffness and difficulty in shifting the pelvis to consecutive steps. He reported difficulty in opening the jaw since childhood. Later, he developed flexed posture of both elbows, became slowly stooped and noticed prominent chin and attributed it to his paternal grandmother. His cognitive functions are normal.
Examination showed short statured boy (height-145 cm, arm span-148 cm, upper segment-70 cm, and lower segment-75 cm), his palpebral fissure was narrow at rest (6 mm gap between upper and lower eyelids) and when attempting to close and open it was becoming still narrower. Patient showed no fatigability. He had bushy eyelashes, flat forehead, receding chin, pursed mouth, high arched palate, and pinched upturned nose. He had a peculiar spasm visible in facial muscles, malocclusion, short neck with neck extensor contractures, mild kyphosis in the thoracolumbar region, bilateral flat foot, and hallux valgus [Figure 1a–c]. His gait showed ankle externally rotated causing a wide base pelvis which moved en bloc and left hip tilted down. His calf muscles were prominent, and there was transient percussion myotonia in the left thenar muscles. The tone, power, reflexes, and sensations were normal in all groups.
Figure 1.
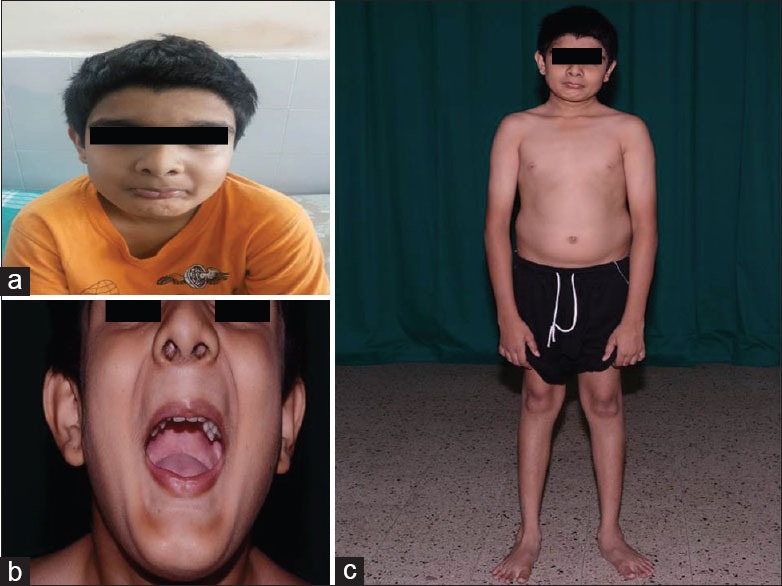
(a) Fixed facial expression, bushy eyebrows, decreased palpebral fissure, low placed ears, pursed lips, micrognathia with myotonic spasm of the chin muscles, (b) difficulty in opening the jaw completely, (c) mild flexion at the elbow, broad base while standing
Investigations
Routine blood chemistry, thyroid function tests, liver and renal function tests, serum lactate, and ammonia were within normal range. His creatine kinase was 294 units. Cardiac evaluation was normal. Tandem mass spectroscopy for metabolic diseases was normal. X-ray of the pelvis and hip was normal [Figure 2]. Nerve conduction studies, both motor and sensory were normal. Needle electromyography was done at 2 sites, right biceps and right quadriceps. The insertional activity and spontaneous activity were abundant and persistent. This consisted of discharges with variation in amplitude and frequency [Figure 3a–c]. Mild needle displacement produced bursts of activity which was associated with dive-bomber sound. When the needle was kept still, the spontaneous discharges continued. Motor unit potential could not be measured properly. There were complex repetitive discharges with uniform morphology. The baseline never became silent. Muscle magnetic resonance imaging and genetics were not done because of financial constraints. Muscle biopsy was done from the left biceps. Paraffin and cryosections showed skeletal muscle tissue with preserved fascicular architecture. The fibers were polygonal with few atrophic and angulated fibers. Focal grouped nuclei and regenerating fibers were seen. Modified Gomori's trichrome stain did not reveal any abnormality. Succinyl dehydrogenase, nicotinamide adenine dehydrogenase, ATPase at pH 9.4 and 4.6 showed type 1 and type 2 grouping. These features were suggestive of denervation with re-innervation. Electron microscopy was not done.
Figure 2.
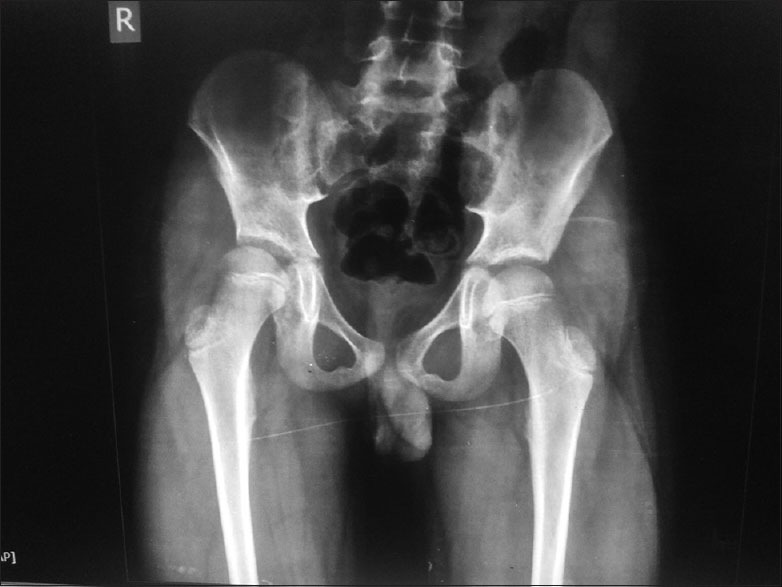
X-ray showing normal pelvic bones in spite of pelvis moving en bloc
Figure 3.
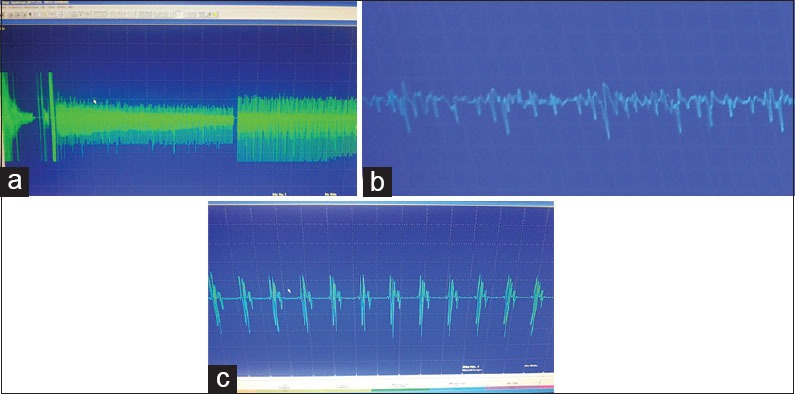
(a) ongoing spontaneous activity trace with Needle insertion (sweep speed 10 ms per division, sensitivity 200 mcV) - shows reduction in the amplitude, (b) continuous discharges showing slight amplitude variation (sweep speed 10 ms per division, sensitivity 200 mcV) (c) the complex repetitive discharges which are stereotyped (sweep speed 10 ms per division, sensitivity 200 mcV)
Discussion and Conclusion
This is a rare congenital myotonic syndrome with distinct clinical and electrophysiological features. This case is being reported for the following reasons. So far, only <100 cases are reported in the literature. Prenatal diagnosis is possible in parents with affected children. The parents need to be warned regarding the risk of malignant hyperthermia during anesthesia. Diagnosis is made primarily by recognition of the clinical phenotype.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Schwartz O, Jampel RS. Congenital blepharophimosis associated with a unique generalized myopathy. Arch Ophthalmol. 1962;68:52–7. doi: 10.1001/archopht.1962.00960030056011. [DOI] [PubMed] [Google Scholar]
- 2.Berardinelli A, Ferrari Ginevra O, Lanzi G. The Schwartz-Jampel syndrome. A minireview. Basic Appl Myol. 1997;7:363–7. [Google Scholar]
- 3. [Last accessed on 2014 Dec 03, 6:00 pm IST]. Available from: http://www.orpha.net/consor4.01/www/cgi-bin/Disease_Search.php?lng=EN and data_id=215 and Disease_Disease_Search_diseaseGroup=schwartz-jampel–syndrome .
- 4.Nicole S, Ben Hamida C, Beighton P, Bakouri S, Belal S, Romero N, et al. Localization of the Schwartz-Jampel syndrome (SJS) locus to chromosome 1p34-p36.1 by homozygosity mapping. Hum Mol Genet. 1995;4:1633–6. doi: 10.1093/hmg/4.9.1633. [DOI] [PubMed] [Google Scholar]
- 5.Cao A, Cianchetti C, Calisti L, de Virgiliis S, Ferreli A, Tangheroni W. Schwartz-Jampel syndrome. Clinical, electrophysiological and histopathological study of a severe variant. J Neurol Sci. 1978;35:175–87. doi: 10.1016/0022-510x(78)90001-1. [DOI] [PubMed] [Google Scholar]
- 6.Fowler WM, Jr, Layzer RB, Taylor RG, Eberle ED, Sims GE, Munsat TL, et al. The Schwartz-Jampel syndrome. Its clinical, physiological and histological expressions. J Neurol Sci. 1974;22:127–46. doi: 10.1016/0022-510x(74)90060-4. [DOI] [PubMed] [Google Scholar]
- 7.Huttenlocher PR, Landwirth J, Hanson V, Gallagher BB, Bensch K. Osteo-chondro-muscular dystrophy. A disorder manifested by multiple skeletal deformities, myotonia, and dystrophic changes in muscle. Pediatrics. 1969;44:945–58. [PubMed] [Google Scholar]
- 8.Topaloglu H, Serdaroglu A, Okan M, Gücüyener K, Topçu M. Improvement of myotonia with carbamazepine in three cases with the Schwartz-Jampel syndrome. Neuropediatrics. 1993;24:232–4. doi: 10.1055/s-2008-1071547. [DOI] [PubMed] [Google Scholar]