

Abstract
Rubinstein–Taybi syndrome (RSTS) is a rare genetic disorder with characteristic morphological anomaly. Our patient was a 4.5-year-old girl came with features like broad thumbs, downward slanting palpebral fissures and mental retardation. Systemic abnormalities such as repeated infection, seizure with developmental delay were also associated with it. She was having head banging behavior abnormal slurring speech, incoordination while transferring things from one hand to other. Galaxy of clinical pictures and magnetic resonance imaging report helped to clinch the diagnosis as a case of “RSTS with corpus callosal agenesis” which to the best of our knowledge has never been reported in past from India.
Keywords: Agenesis, corpus callosum, Rubinstein–Taybi syndrome
Introduction
Rubinstein–Taybi syndrome (RSTS) or broad thumb-hallux syndrome was initially described by Michail et al. in 1957. Craniofacial features and global mental retardation are characteristic. Individuals with RSTS rarely reproduce. Inheritance is autosomal dominant. There is an increased risk of developing meningioma, other brain tumors and leukemia.[1] Rarely, RSTS is associated with agenesis of corpus callosum (ACC).[2] The frequent clinical findings in patients with ACC are mental retardation, visual problems, speech delay, seizures, and feeding problems[3] (in our patient it is seizure). RSTS with ACC is never reported from our country before.
Case Report
A 4 years and 6 months old female child presented to pediatric outdoor with a chief complaint of fever for 1-month with vomiting and lethargy. She had no history of hemoptysis, rapid weight loss, prolonged headache, or severe vomiting. The child was suffering from repeated attacks of fever and respiratory tract infection requiring hospitalization from infancy. On day seven of life, she had seizure and was treated in local hospital. That was followed by head banging movements which persisted for 1-year. She was on antiepileptic treatment for 2 years. Developmentally there was significant language and motor delay. She was immunized according to age. She was born by normal vaginal delivery with no significant perinatal abnormality. She had significant morphological abnormality with hypertelorism, beaked nose, downward slanting, high arched palate, broad thumb and big toe, enlarged palmer web, dental malalignment, hyperextensive joints, head circumference less than 50th percentile etc. [Figures 1 and 2]. All vitals were stable except mild fever which was measured to be 100.5 F. Child had significant mental retardation, abnormal slurring speech, incoordination while transferring things from one hand to other and some behavioral anomaly. Rest neurological examinations were normal. Mild hepatosplenomegaly was there. Liver measured to be 3 cm and spleen 2 cm. All other systemic examinations were normal. Routine blood investigation revealed Hb to be 7.4 mg/dl, malarial parasite antigen immuno-chromatographic test (optimal-antigen) was negative. Widal test and Typhidot were positive. Erythrocyte sedimentation rate was 15 on 1st hr. All other blood pictures were normal. Electrocardiogram showed no cardiac defect. Magnetic resonance imaging showed ACC with polymicrogyria [Figure 3]. Basing on all clinical features and morphological abnormality, the child was diagnosed as RSTS with ACC with enteric fever. She was treated by intravenous antibiotics, and the fever disappeared on 4th day of treatment.
Figure 1.
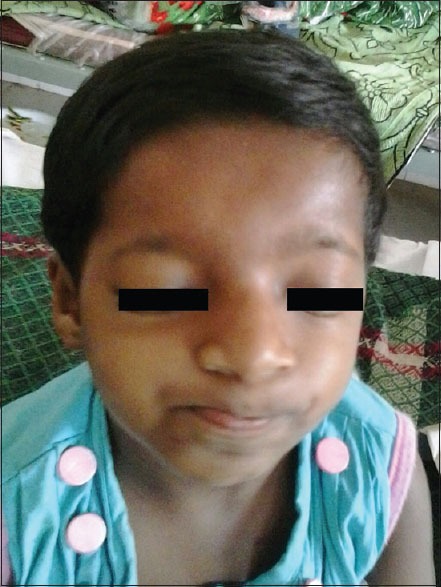
Facial profile of Rubinstein–Taybi syndrome
Figure 2.

Classical broad thumb
Figure 3.

Magnetic resonance imaging showing agenesis of corpus callosum and polymicrogyria
Discussion
Rubinstein-Taybi syndrome is a very rare syndrome. The incidence of disease is 1:300,000 at birth.[4] Less than 30 cases have been reported so far in the Indian literature.[5] RSTS is the only disorder known to be associated with germline mutations in CREB-binding protein region (CREBBP) and EP300.[6] Cause is unclear but microdeletion of 16p 13.3 region in the CREBBP has been found in some patients, suggesting it to be the cause of the syndrome.[7] CREBBP is a transcription coactivator and acts as a potent histone acetyl transferase. The empirical recurrence risk after an earlier child with RSTS is 0.1%. It produces galaxy of clinical features among them the craniofacial (down slanting palpebral fissures, high arched palate, and beaked nose with the columella extending below the nares, grimacing smile, and talon cusps)[6] and limb symptoms are the most common. Along with it ophthalmic features like strabismus, refractory errors, ptosis, nasolacrimal duct obstruction, cataracts, coloboma, nystagmus, glaucoma, and corneal abnormalities are not rare. Congenital heart disease is found in 1/3rd of cases. Renal abnormalities are very common and almost all boys have undescended testes. Orthopedic issues include dislocated patellas, lax joints, spine curvatures, slipped capital femoral epiphysis, and cervical vertebral abnormalities.[8] Keloids, hirsutism may occur with only minimal trauma to the skin.[2] Dental problems include crowding of teeth, malocclusion, multiple caries, natal teeth, and talon cusps on the upper incisors of the secondary dentition. Tumors reported in individuals with RSTS include meningioma, pilomatrixoma, rhabdomyosarcoma, pheochromocytoma, neuroblastoma, medulloblastoma, oligodendroglioma, leiomyosarcoma, and leukemia.[9] Speech delay occurs in 90% of children, and some remain largely nonverbal. Short attention span, decreased tolerance for noise and crowds, impulsivity, and moodiness are frequently observed. Other abnormal behaviors included attention problems, hyperactivity, self-injurious, and aggressive behaviors.[6] The association of corpus callosum and RSTS is very rare and scantily reported.[2,3] In a review of the embryology of the corpus callosum, Dobyns suggested that several difierent mechanisms can result in ACC. Two primary or “true” types of ACC and two secondary types have been recognized. True ACC include (1) Defects where axons form, but are unable to cross the midline because of absence of the massa commissuralis and lead large aberrant longitudinal fiber bundles known as Probst bundles along the medial hemispheric walls; and (2) defects in which the commissural axons or their parent cell bodies failed to form in the cerebral cortex. The former, most common type of ACC occurs in all ACC syndromes in which Probst bundles are seen.[10] ACC may occasionally occur without any apparent associated abnormalities by clinical examination or currently available neuroimaging studies. This is called isolated or primary ACC. The most frequent clinical findings in patients with ACC are mental retardation (60%), visual problems (33%), speech delay (29%), seizures (25%), and feeding problems (20%).[11] Furthermore, people with primary AgCC may display a variety of other social, attentional, and behavioral symptoms that can resemble those of certain psychiatric disorders.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet. 1995;56:112–5. doi: 10.1002/ajmg.1320560125. [DOI] [PubMed] [Google Scholar]
- 2.Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, et al. Agenesis of the corpus callosum: Genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8:287–99. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- 3.Kniffin CL, McKusick VA. Corpus Callosum, Agenesis of, Clinical Synopsis OMIM entry – % 217990, SNOMEDCT: 5102002 ICD10CM: Q04. [Last updated 2011 Jun 06]. Available from: http://www.omim.org/entry/217990 . Creation date-victor A-McKusick-6/3/1986.
- 4.Morales-Chávez MC. Dental management of a patient with Rubinstein-Taybi syndrome. Spec Care Dentist. 2010;30:124–6. doi: 10.1111/j.1754-4505.2010.00137.x. [DOI] [PubMed] [Google Scholar]
- 5.Bansal S, Relhan V, Garg VK. Rubinstein-Taybi syndrome: A report of two siblings with unreported cutaneous stigmata. Indian J Dermatol Venereol Leprol. 2013;79:714–7. doi: 10.4103/0378-6323.116751. [DOI] [PubMed] [Google Scholar]
- 6.Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CRt, Fong CT, et al. Rubinstein-Taybi syndrome. [Last update 2014 Aug 07];GeneReviews® [Internet] Initial Posting. 2002 Aug 30; [Google Scholar]
- 7.Petrij F, Dauwerse HG, Blough RI, Giles RH, van der Smagt JJ, Wallerstein R, et al. Diagnostic analysis of the Rubinstein-Taybi syndrome: Five cosmids should be used for microdeletion detection and low number of protein truncating mutations. J Med Genet. 2000;37:168–76. doi: 10.1136/jmg.37.3.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto T, Kurosawa K, Masuno M, Okuzumi S, Kondo S, Miyama S, et al. Congenital anomaly of cervical vertebrae is a major complication of Rubinstein-Taybi syndrome. Am J Med Genet A. 2005;135:130–3. doi: 10.1002/ajmg.a.30708. [DOI] [PubMed] [Google Scholar]
- 9.Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: Clinical and molecular overview. Expert Rev Mol Med. 2007;9:1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- 10.Dobyns WB. Absence makes the search grow longer. Am J Hum Genet. 1996;58:7–16. [PMC free article] [PubMed] [Google Scholar]
- 11.Schell-Apacik CC, Wagner K, Bihler M, Ertl-Wagner B, Heinrich U, Klopocki E, et al. Agenesis and dysgenesis of the corpus callosum: Clinical, genetic and neuroimaging findings in a series of 41 patients. Am J Med Genet A. 2008;146A:2501–11. doi: 10.1002/ajmg.a.32476. [DOI] [PMC free article] [PubMed] [Google Scholar]