

Abstract
Introduction:
About one-half of children with Lennox–Gastaut syndrome (LGS) have history of birth hypoxia or other perinatal event but the knowledge about clinical, radiological profile and severity of epilepsy in these children as compared to those without a perinatal event is not known.
Materials and Methods:
Thirty-one children with LGS were enrolled in this study and divided into two groups: One group with the perinatal event and other group without evidence of the perinatal event. We hypothesized that LGS with the perinatal event will have an early age of onset of LGS, more motor deficits and abnormal brain magnetic resonance imaging (MRI) and more severe epilepsy.
Results:
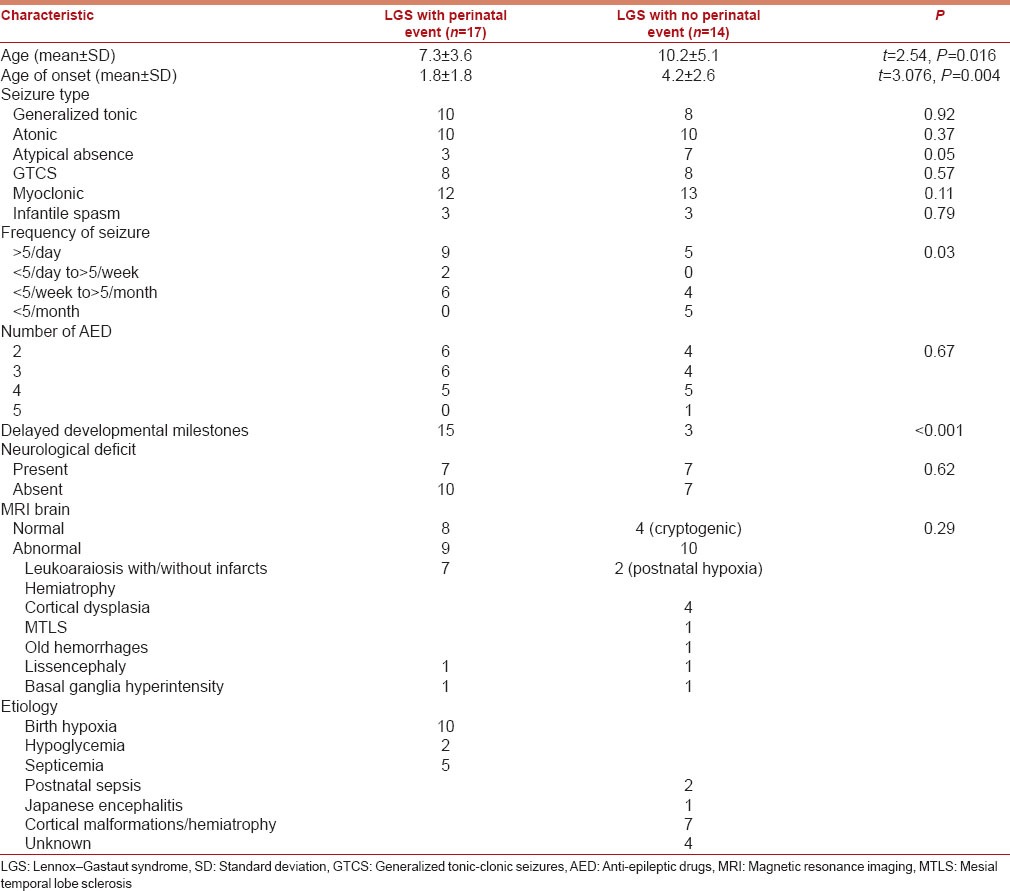
There were 17 children in the perinatal event group and 14 in the other group. The mean age of onset of illness was significantly earlier in the perinatal event group (P < 0.05). More children in the perinatal event group had delayed milestones (P < 0.05), had higher seizure frequency (P < 0.05) however; there was no significant difference in number of anti-epileptic drugs consumed, motor deficits or MRI abnormalities.
Conclusion:
LGS children with the perinatal event have more severe epilepsy with early onset of disease and delayed milestones. History of perinatal insult in these children may help in predicting prognosis in LGS.
Keywords: Atonic seizure, Lennox–Gastaut syndrome, perinatal hypoxia, slow-spike-wave discharges
Introduction
Lennox–Gastaut syndrome (LGS) belongs to a group of severe childhood epileptic encephalopathy characterized by the symptomatic triad of several seizure types, characteristic electroencephalographic (EEG) abnormalities and mental retardation and/or personality changes.[1,2,3] LGS is estimated to comprise 1–4% of all childhood epilepsies.[4,5] These children mainly suffer from the generalized tonic, atonic, and atypical absence seizures. However, many children eventually develop generalized tonic-clonic seizures (GTCS), myoclonic jerks, and partial/complex partial seizures. The typical EEG abnormality seen in these children includes generalized interictal slow-spike-wave discharges (<3 Hz), fast rhythmic 10 Hz burst (with tonic seizures) and interictal multi-focal spike/sharp wave discharges. Nearly two-third of children with LGS are classified into symptomatic (“structural-metabolic”) group and one-third into cryptogenic (“unknown cause”) group.[6,7] Structural-metabolic group comprise of children with cortical malformations, tuberous sclerosis, hypoxic-ischemic encephalopathy, and rarely with inborn errors of metabolism. Those with normal magnetic resonance imaging (MRI) mostly classify into cryptogenic group. It is estimated that about half of all LGS patients and 41% of West syndrome patients who evolve into LGS have history of perinatal hypoxia.[8,9] Despite a well-agreed classification of LGS into “structural-metabolic” and “unknown cause” based on presumed etiology, both these groups hardly differ from each other in terms of seizure types, seizure frequency, cognitive functions, EEG abnormalities or in respect to overall prognosis.[10,11,12] However, despite a strong association of perinatal hypoxia and LGS, there is little knowledge regarding any difference between LGS children with and without perinatal hypoxia or other perinatal events in terms of demographic, clinical, and radiological characteristics.
Since, children with birth hypoxia or other perinatal insults have higher chance of focal neurological deficits/structural brain abnormalities, higher prevalence of West syndrome, therefore, we hypothesize that LGS children with a perinatal hypoxia or other perinatal event will have; (1) a higher chance of a preceding West syndrome, (2) early age of onset of LGS,(3) a higher chance to acquire focal neurological deficits and structural brain abnormality on MRI.
Materials and Methods
This was a cross-sectional study conducted in a tertiary health care facility in northern India from April 2011 to January 2013. All consecutive children (up to 18 years) diagnosed with LGS in our neurology outpatient department were included in the study. Diagnosis of LGS was considered in children having:
Three or more seizure types with at least one of the following: Tonic seizures, atonic seizures or atypical absence seizures
At least one EEG showing generalized slow-spike-wave of <3 Hz activity or sleep EEG showing rhythmic fast (10 Hz) activity
Mental subnormality.
Parents or guardian of children gave their written consent for inclusion in the study. Institutional ethics committee approved the study.
Detailed record of age of onset of seizures, frequency/type of seizures, number of anti-epileptic drugs (AEDs) being consumed, compliance to drugs, history of febrile seizures, febrile encephalopathy and its squeal was obtained from parents. History regarding antenatal, perinatal, and postnatal period was inquired from mother. Medical records were checked for any antenatal problems, perinatal insults like birth asphyxia, Apgar score, pathological jaundice, stay in a neonatal intensive care unit and cause of stay thereof. A detailed account of social, motor, language, and other cognitive milestones like toilet training, behavior with other children, school performance, hyperkinetic behavior, etc., were also noted. Complete general and systemic examination was performed and records of any neurocutaneous markers, facial dysmorphism, and musculoskeletal abnormality were made. A detailed neurological examination was also performed.
Perinatal hypoxia was defined as a “failure to initiate and sustain breathing at birth”. All neonates that fulfilled any one of the two of criteria in all three group were considered as having perinatal hypoxia: (1) Signs of intrauterine asphyxia, e.g., pathologic cardiotocogram (<80 beats/min, limited beat-to-beat variability, late deceleration), meconium stained amniotic fluid; (2) signs of perinatal asphyxia, e.g., 5-min Apgar score ≤5, cord pH ≤7.1, base deficit ≥10 mmol/L; and (3) signs of postpartum encephalopathy starting within the first 48 h, e.g., decreased muscle tone, pathologic spontaneous movements, lethargy, coma, or seizures.[13] Those with seizures, unconsciousness, febrile encephalopathy or any other brain insult up to 28 days after birth were also included into the group of children with perinatal insult.
A-21-channel EEG record was obtained for all children using 10–20 international system of electrode placement. Every child had a MRI of brain that was evaluated independently by two radiologists for any structural brain abnormality.
Statistical analysis
All children with LGS were divided into two groups. First group included children with a history of the perinatal event whereas second group had children without evidence of the perinatal event. Independent and continuous variable between the two groups were compared using Student's t-test and Chi-square test.
Results
About 500 children diagnosed with epilepsy were enrolled in the neurology out-door of our institute during the study period. Thirty-five children were diagnosed as LGS as per above mentioned diagnostic criteria. Four children were excluded from the study due to unreliable birth history and lack of medical records. Thirty-one patients of LGS were enrolled in this study. Demographic details, clinical, EEG, and MRI features of all children are summarized in Table 1. Median age of the patients was 8 years. Twenty-five patients were male. Mean age of onset of seizures was 8.757 ± 2.05 years (1–11 years). Mean duration of the illness (duration from the onset of seizures to age at the time of inclusion in the study) was 5.96 ± 3.27 years.
Table 1.
Clinical, radiological, and EEG characteristics of 31 LGS children

Most common seizure type was myoclonic jerks (n = 25, 80.6%) followed by atonic seizures (n = 20, 64.5%), generalized tonic seizures (n = 18, 58%), GTCS (n = 16, 51.6%), atypical absence (n = 10, 32.3%), and complex partial seizures (n = 11, 35.5%). Preceding West syndrome was seen only in 6 (19.4%) patients. None of these children had preceding EME or Ohtahara syndrome. To ascertain seizure frequency parents kept a month-long record of seizures of their children and every seizure type was counted. Fourteen children had more than five seizures per day, 2 children had more than five seizures per month, 10 children had more than five seizures per week, and 5 children had <5 seizures per month. Ten children each were taking 2, 3, and 4 AEDs, respectively, and one child was taking 5 AEDs at the time of inclusion in the study.
Spastic diplegia was present in 4 patients, spastic quadriparesis in 2 patients, only spasticity without weakness in lower limbs in 4 patients, hemiparesis in 2, extrapyramidal features in 1, and generalized hypotonia was present in 1 patient.
Magnetic resonance imaging brain abnormality was seen in 19 (60%) children. All children showed slow-spike-wave discharges (1.5–2.5 Hz) that were generalized in 29 and 2 showed predominance on one side. Only one child had 10 Hz fast burst during sleep. All children had multifocal spikes and sharp waves. Patients with West syndrome had initial features of hypsarrhythmia that later changed to generalized slow-spike-wave discharge.
Demographic, clinical and MRI features were compared between children with LGS and a perinatal event group with LGS without any perinatal event group [Table 2]. There were 17 children in the perinatal event group and 14 in the other group. Only 2 children had a history of premature birth but without any perinatal insult. Most common underlying cause among LGS without perinatal event group was cortical malformations in 7 children. Other etiologies were postnatal septicemia in 2, Japanese encephalitis in 1 and unknown cause (cryptogenic) in 4 children. In the perinatal event group, 10 children had perinatal hypoxia, five had septicemia, and two had hypoglycemia. The mean age of onset of seizures was significantly earlier in the perinatal event group (1.8 ± 1.8 years vs. 4.2 ± 2.6 years) (P = 0.004). More children in the perinatal event group had delayed milestones (13 vs. 5) (P ≤ 0.001). Perinatal event group had a higher frequency of seizure (P = 0.03). More children without a perinatal event had atypical absence seizures as compared to those with a perinatal event (P = 0.05) However, there was no significant difference in other seizure types, number of AEDs consumed, motor deficits or number of children with abnormal brain MRI between the either groups.
Table 2.
Comparison of LGS children with and without perinatal hypoxic insult
Discussion
This study partially supports our hypothesis. More than 50% of children with LGS had a history of a perinatal event in our study that conforms to previous studies.[14] LGS with perinatal event group showed early age of onset of LGS, had more frequent seizures and exhibited delayed milestones in a higher proportion of children as compared to LGS without perinatal event. In our study, LGS children with a perinatal event were younger than those without a perinatal event. This was possibly because of earlier onset of seizures and delayed milestones in these children that necessitated medical advice at a younger age.
The pathogenesis of LGS is not known. The localization of epileptogenic regions of the brain that produces different seizure types and the underlying cause of brain pathology is not known.[15] Various focal brain abnormalities associated with LGS like cortical dysplasia, hypoxic-ischemic changes, hypoglycemia, and encephalitis related abnormalities fail to explain satisfactorily the appearance of different types of seizures and mental decline in these children. It is also interesting to note that the LGS children without any underlying cause and with normal brain MRI have similar phenotype and similar prognosis as children with symptomatic LGS.[16,17] Therefore, the classification of LGS into “symptomatic” and “cryptogenic” is not clinically useful despite having a valid basis of classification based on presumed underlying etiology of LGS. Since, a large proportion of children with LGS suffer from perinatal hypoxia; classifying LGS into groups with and without the perinatal event is useful for the clinician in predicting prognosis.
Most other variables that we studied in our cohort like abnormal MRI, motor deficits, number of anti-epileptics being consumed and seizure types were similar in either group. This reinforces the fact that LGS children with perinatal hypoxia have similar phenotype as other symptomatic and cryptogenic LGS. However, significant perinatal brain insult for some unknown reason may unmask/de novo produce this syndrome at an earlier age and depending upon the severity of insult may add delayed milestones to this syndrome. Higher seizure frequency in these children could be due to kindling effect due to an earlier age of onset of epilepsy.
More children in the group “without a perinatal event” suffered atypical absences. This was possibly because absences are more readily noticeable and reported by parents in grown-up children and are difficult to ascertain in infancy and early childhood.
Our study had certain limitations. We had a small cohort of LGS children. Owing to the different age group of children, we could not use a uniform scale to measure the mental status of children and, therefore, could not compare the degree of cognitive dysfunction in either group. We could perform only a single 1-month follow-up in our children. We also had a higher frequency of myoclonic jerks in our cohort. This possibly occurred due to erroneous recording of flexor/extensor spasms by parents as myoclonic jerks in some children.
Conclusion
Lennox–Gastaut syndrome children with a history of perinatal hypoxia or other perinatal event have earlier age of onset of seizures; have delayed milestones and a higher seizure frequency as compared to LGS children without a perinatal event. History of perinatal hypoxia or other significant perinatal insult in children with LGS may help in predicting their prognosis and planning a more aggressive management.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Engel J., Jr ILAE classification of epilepsy syndromes. Epilepsy Res. 2006;70(Suppl 1):S5–10. doi: 10.1016/j.eplepsyres.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Beaumanoir A. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood, and Adoloscence. London: John Libbey; 1985. pp. 89–99. [Google Scholar]
- 3.Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989;30:389–99. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- 4.Heiskala H. Community-based study of Lennox-Gastaut syndrome. Epilepsia. 1997;38:526–31. doi: 10.1111/j.1528-1157.1997.tb01136.x. [DOI] [PubMed] [Google Scholar]
- 5.Trevathan E, Murphy CC, Yeargin-Allsopp M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia. 1997;38:1283–8. doi: 10.1111/j.1528-1157.1997.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 6.Arzimanoglou A, French J, Blume WT, Cross JH, Ernst JP, Feucht M, et al. Lennox-Gastaut syndrome: A consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol. 2009;8:82–93. doi: 10.1016/S1474-4422(08)70292-8. [DOI] [PubMed] [Google Scholar]
- 7.Markand ON. Slow spike-wave activity in EEG and associated clinical features: Often called ‘Lennox’ or “Lennox-Gastaut’ syndrome. Neurology. 1977;27:746–57. doi: 10.1212/wnl.27.8.746. [DOI] [PubMed] [Google Scholar]
- 8.Yamatogi Y, Ohtahara S. Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev. 2002;24:13–23. doi: 10.1016/s0387-7604(01)00392-8. [DOI] [PubMed] [Google Scholar]
- 9.Weinmann HM. Lennox Gastaut syndrome and its relationship to infantile spasms (West syndrome) In: Neidermeyer E, Degen R, editors. The Lennox Gastaut Syndrome. NewYork: Alan R Liss; 1988. pp. 301–16. [Google Scholar]
- 10.Goldsmith IL, Zupanc ML, Buchhalter JR. Long-term seizure outcome in 74 patients with Lennox-Gastaut syndrome: Effects of incorporating MRI head imaging in defining the cryptogenic subgroup. Epilepsia. 2000;41:395–9. doi: 10.1111/j.1528-1157.2000.tb00179.x. [DOI] [PubMed] [Google Scholar]
- 11.Oguni H, Hayashi K, Osawa M. Long-term prognosis of Lennox-Gastaut syndrome. Epilepsia. 1996;37(Suppl 3):44–7. doi: 10.1111/j.1528-1157.1996.tb01820.x. [DOI] [PubMed] [Google Scholar]
- 12.Chevrie JJ, Aicardi J. Childhood epileptic encephalopathy with slow spike-wave. A statistical study of 80 cases. Epilepsia. 1972;13:259–71. doi: 10.1111/j.1528-1157.1972.tb05260.x. [DOI] [PubMed] [Google Scholar]
- 13.Ersdal HL, Mduma E, Svensen E, Perlman J. Birth asphyxia: A major cause of early neonatal mortality in a Tanzanian rural hospital. Pediatrics. 2012;129:e1238–43. doi: 10.1542/peds.2011-3134. [DOI] [PubMed] [Google Scholar]
- 14.Ren LK, Wu LW, Jin LR, Gao W, Shao XQ. Characteristics of clinical manifestations and EEG of Lennox-Gastaut syndrome. Zhonghua Er Ke Za Zhi. 2003;41:7–10. [PubMed] [Google Scholar]
- 15.Blume WT. Pathogenesis of Lennox-Gastaut syndrome: Considerations and hypotheses. Epileptic Disord. 2001;3:183–96. [PubMed] [Google Scholar]
- 16.Gastraut H, Roger J, Soulayrol R, Tassinari CA, Régis H, Dravet C, et al. Childhood epileptic encephalopathy with diffuse slow spike-waves (otherwise known as “petit mal variant”) or Lennox syndrome. Epilepsia. 1966;7:139–79. doi: 10.1111/j.1528-1167.1966.tb06263.x. [DOI] [PubMed] [Google Scholar]
- 17.Neidermeyer E. The Lennox Gastaut syndrome: A severe type of childhood epilepsy. J Neurol. 1969;195:263–82. doi: 10.1007/BF00242459. [DOI] [PubMed] [Google Scholar]