

Abstract
5'-Ether derivatives of the potent adenosine agonist N6-cyclopentyladenosine (CPA) were designed as “caged” ligands for the activation of A1-adenosine receptors following in situ photolysis. The synthesis involved a 2',3'-diol protection scheme using the acid labile ethoxymethynyl group. Generation of CPA was demonstrated chromatographically and in a bioassay measuring the inhibition of synaptic potentials in the rat hippocampus.
Introduction
Adenosine acts as a neuromodulator in the CNS1 and as a regulator of the activity of other organs, such as the heart2 and kidneys,3 generally as a protective depressant of the organ's activity in response to excessive oxygen demand and/or diminished oxygen supply.4 In the central nervous system, adenosine agonists act to depress neuronal firing and the release of stimulatory neurotransmitters, such as acetylcholine5 and glutamic acid.6 The anticonvulsant effects of adenosine agonists in the hippocampus have been described.7 A difficulty in the use of adenosine agonists in physiological systems is the apparent lag time between the application of the drug and the effect observed. This phenomenon has been ascribed to limited diffusion8,9 of the normally hydrophilic adenosine derivative to the receptor site or to a mechanistic delay, in which adenosine receptors require a postulated biochemical activation.10
A technique for examining the fast kinetics of neurotransmitter action is that of “caged” probes,11 in which the biologically active form of a molecule is generated in situ from an inactive precursor through a photochemical deblocking process. This technique has been developed for nucleotides and other neurotransmitters and biochemical messengers,11 such as ATP, 3',5'-cyclic AMP, inositol-1,4,5-triphosphate, γ-aminobutyric acid,12 and glutamic acid.12 The photolabile linkages used previously and in this study are based on the 2-nitrobenzyloxy group, introduced by Patchornik and coworkers.13,14
Materials and Methods
Chemistry
2-Nitrobenzylbromide was purchased from Lancaster Synthesis (Windham, NH). N6-Cyclopentyladenosine (CPA) and 1-(bromomethyl)-4,5-dimethoxy-2-nitrobenzene (NV-Br) were synthesized as reported.13b, 17 Analytical TLC plates and silica gel (230–400 mesh) were purchased from VWR. 1NMR spectra in CDCI3 or DMSO-d6 were obtained at 300 MHz on a VARIAN GEMINI-300. Mass Spectra were obtained using a Finnigan 10150 for CI and using a Jeol JMS-SX102 for FAB ionization. Elemental analyses were by Galbraith Labs (Knoxville, TN).
N6-Cyclopentyl-2'-3'-O-ethoxymethylene-adenosine. 2
CPA, 1, (500 mg, 1.5 mmol) and trichloroacetic acid (500 mg, 3 mmol) were dissolved in dioxane (25 ml), and the mixture was stirred at room temperature over 4Å molecular sieves. Triethylorthoformate was added (2 ml, 12 mmol), and the solution heated at 60°C for 45 min under a nitrogen atmosphere. Sodium bicarbonate (1 g, 12 mmol) was added at 0° C, and after 1.5 h of vigorous shaking the mixture was filtered and the solvent evaporated. The residue was purified by flash column chromatography on silica gel with chloroform:methanol, 98:2, by vol, as eluent. Evaporation of the solvent from the pure fraction afforded 480 mg of 2 as an oil in quantitative yield.
1H-NMR δ (DMSO-d6): 1.1 & 1.2 (t, 3H, CH3 endo/exo, J = 7Hz), 1.6, 1.7 & 1.95 (m, 8H, Cp), 3.5–3.7 (m, 4H, CH2O & 5'-H), 4.2 & 4.3 (m, 1H, 4'-H), 4.5 (m, 1H, CHN), 4.95 & 5.05 (dd, 1H, 3'-H endo/exo, J3', 4' = 3.5 Hz, J3', 2'' = 7.1 Hz), 5.40 & 5.46 (dd, 1H, 2'-H endo/exo, J2', 1' = 3.1 Hz), 6.17 & 6.21 (s, 1H, O2CH endo/exo), 6.28 (d, 1H, 1'-H), 7.78 & 8.2 (Is, 1H endo/exo, NH), 8.32 & 8.4 (s & d, 2H, 2-H, 8-H). Anal. calcd. for C18H25N5O5.0.5CHCl3: C, 49.17; H,.5.64; N, 15.05. Found: C, 49.26; H,.5.70; N, 15.52. MS (CI-NH3) 392 M+1, 204 Base+2. High resolution MS (FAB/ NOBA matrix): m/e 392.1945, M+1, 392.1934 calcd. for C18H26N5O5
N6-Cyclopentyl-5'-O-(2-nitrobenzyl)-2'-3'-O-ethoxymethylene-adenosine, 3a
Sodium hydride (15 mg of 50% oil dispersion, 0.26 mmol) was washed with pentane and was dispersed in 2 ml of dry DMF. After cooling the suspension to 0°C compound 2 (50 mg, 0.13 mmol) in 2.5 ml of DMF was added. After gas generation had ceased (45 min), 2-nitrobenzylbromide (70 mg, 0.26 mmol) was added in 0.5 ml of DMF at 0° C in the dark, and the mixture was stirred at room temperature overnight. The reaction was quenched with ethanol, and diluted with H2O and then extracted 3 times with Et2O. The combined Et2O layers were dried and after evaporation the residue was purified by flash chromatography with hexane:EtOAc, 1:1 to 1:0, by vol, to give 19 mg of 3a (28%) as an oil, and 25 mg of starting material 2 (50%) was recovered.
1H-NMR δ (CDCI3): 1.3 (m, 3H, CH3), 1.5–1.8 & 2.(m, 8H, Cp), 3.7 (m, 2H, CH3CH2O), 4.39 (dd, 1H, 5'-H, J gem = 11.5 Hz, J4'–5' = 7 Hz,) 4.47 (dd, 1H, 5'-H, J4'–5' = 4.5 Hz), 4.62 (m, 1H, 4'-H), 5.10 (m, 3H, 3'-H & PhCH2O), 5.59 (dd, 1H, 2'H, J2', 3' = 7.1 Hz, J2', 1' = 2.3 Hz), 5.70 (m, 1H, CHN), 6.04 & 6.06 (s, 1H, O2CH endo/exo), 6.20 & 6.22 (d, 1H, 1'-H endo/exo), 7.19 (d, 1H, o-H-phenyl, J = 8 Hz), 7.44 (t, 1H, p-H-phenyl), 7.56 (dt, 1H, m-H-phenyl, J = 1 Hz), 7.80 & 7.84 (s, 1H, 8-H exo/endo), 8.45 (dd, 1H, m-H-phenyl) 8.30 & 8.34 (br s, 1H, 2-H exo/endo). Anal. calcd. for C25H30N6O7: C, 57.03; H,.5.74. Found: C, 57.18; H,.5.65. MS (CI-NH3) 527 M+1, 497 MH-ethane, 434 MH2-N02Benzyl, 204 Base+2.
N6-Cyclopentyl-5'-O-(2-nitrobenzyl)-adenosina. 4a
Compound 3a (19 mg, 0.04 mmol) was dissolved in 5 ml of chloroform. Trifluoroacetic acid (100 μl) was added at 0°C. After stirring overnight at 8°C an equal amount of TFA (100 μl) was added and the mixture was stirred for an additional 12 h. The solvent was then coevaporated several times with ethanol and the residue was purified by flash chromatography using ethyl acetate followed by ethyl acetate:methanol, 98:2, by vol. After concentration of the desired fraction, 6 mg of the product 4a was isolated as an oil (56%). This compound applied t a TLC plate upon irradiation with 254 nm light for 1 hour was totally converted to CPA. 1H-NMR δ (CDCI3): 1.5 –1.8 & 2.1 (m, 8H, Cp), 3.84 (m, 2H, 5'-H), 4.48 (m, 1H, 3'-H), 4.50 (m, 1H, 4'-H), 4.56 (m, 1H, 2'-H), 5.94 (d, 1H, J1',2' = 5.3 Hz), 7.44 (m, 3H, Harom), 7.98 (s, 1H, 8-H), 8.2 (d, 1H, m-H-phenyl), 8.30 (br s, 1H, 2-H). High resolution MS (FAB/ NOBA matrix): m/e 471.1982, M+1, 471.1992 calcd. for C22H27N6O6.
N6-Cyclopentyl-5'-O-(6-nitroveratryl)-adenosine. 4b
The procedures described above for 3a and 4a were followed starting from NV-Br. The overall yield starting from CPA was 9%, and the product was obtained as a yellow oil. NMR were consistent for 3b and 4b. High resolution MS (FAB/ NOBA matrix) 3b : m/e 587.2478, M+1, 587.2478 calcd. for C27H35N6O9, 4b : m/e 531.2170, M+1, 531.2203 calcd. for C24H31N6O8.
Biological assays
Hippocampal slices were prepared from adults rats (200–300 g) as previously described.15 Field excitatory postsynaptic potentials (fEPSPs) were recorded in the stratum radiatum of the CA1 region in response to stimulation of Schaffer collateral /commisural afferents. Slices were superfused continuously with artificial cerebrospinal fluid (ACSF)15 and fEPSPs were sampled every 15 seconds.
The amplitude of fEPSPs responses was measured on-line and plotted over the time course of each experiment. The magnitude of the inhibitory effect of a compound on fEPSP amplitude was expressed as the percentage inhibition, according to the formula: Percentage inhibition = [(Amplitude in ACSF only - Amplitude in drug) ÷ Amplitude in ACSF only] × 100.
The following compounds were added, alone or in combination, to the ACSF, and the samples were superfused for 30–40 minutes over the slices: CPA (10, 50 and 100 nM), non irradiated 4a (50 and 100 nM), irradiated 4a (10, 50 and 100 nM), non irradiated 4b (50 nM), irradiated 4b (10, 20 and 50 nM), and theophylline (200 μM). For the irradiation of compounds 4a and 4b, a 10 μM stock solution was placed under a Spectroline UV light source (254 nm) for 10 min. The 10 min period was selected because of the relatively weak source of UV irradiation. The stock solution was then added to the ACSF to achieve the desired concentration, and inhibition was measured.
Results
The structure activity relationships for adenosine analogs have been explored in great detail.16 In general, monosubstitution by alkyl,17 cycloalkyl,18 or aryl19 groups as N6-amino substituents has been associated with agonists of high potency and selectivity for A1-receptors. A2-selective agonists20 have been synthesized through substitutions at the 2-position of the purine ring and/or with N-(small alkyl)carboxamide substitution at the 5'-position. For A1-selective compounds, such as N6-cyclopentyladenosine (CPA), the integrity of the ribose ring is generally important for agonist activity, and certainly at the 5'-position there are severe steric constraints. Thus, the 5'-hydroxyl was selected as the site for the incorporation of a photolabile blocking group.
A. Synthesis
The photolabile groups used included the 2-nitrobenzyl group13 (NB) and the 6-nitroveratryl group21 (NV), shown previously to cleave upon ultraviolet irradiation. 5'-NB-CPA, 4a, and 5'-NV-CPA, 4b, were synthesized using an alcohol protection scheme involving the acid labile 2,'3'-ethoxymethynyl group introduced by reaction of 1 with triethylorthoformate. The large excess of reagent required to drive the reaction to completion also caused the formation of an undesired product bearing a 5'-orthoester. This 5'-blocked by-product was reconverted to the desired compound during the purification on silica gel, giving the protected derivative, 2, in one step with an 86% yield (Scheme 1). Attachment of the photolabile blocking group required O-alkylation of 2 which was achieved through treatment with 2-fold excess of sodium hydride in dimethylformamide at 0° C followed by reaction with an excess of the corresponding 2-nitrobenzyl bromide. The modest yield generally obtained21 (~30%) in this step was not improved by changing the nature of the leaving group (-CI, -I, -OTf). Isolation of the stilbene derivative, 5, in both cases may suggest that a carbene formed by α-elimination is involved in competing side reactions.
SCHEME 1.

5
The final deprotection to give 4a–b was carried out under mild conditions (4% trifluoroacetic acid in CHCI3). The final product is isolated by flash chromatography on silica gei in yields of 50–60 % (Scheme).
B. Bioassay for activity
Adenosine derivatives are potent inhibitors of synaptic potentials in the hippocampal formation, an action mediated by A1-receptors.22 Synaptic responses (fEPSPs) were recorded in the CA1 region of in vitro hippocampal slices, and the actions of the following adenosine derivatives were tested: CPA, 4a (irradiated and non-irradiated), and 4b (irradiated and non-irradiated). The adenosine antagonist theophylline blocked the inhibitory effects of these derivatives (shown in Figure, for 4a). All data shown here for irradiated compounds are from a ten minute period of irradiation. Preliminary data with the the same, simple light source source indicate that a 1 min period of irradiation of compound 4a (50 nM) resulted in an approx. 50% inhibition of fEPSPs. Non-irradiated 4a and 4b elicited less than 10% inhibition of fEPSPs at concentrations up to 50 nM. At a concentration of 100 nM non-irradiated 4a inhibited by approximately 10% (Figure).
Figure.
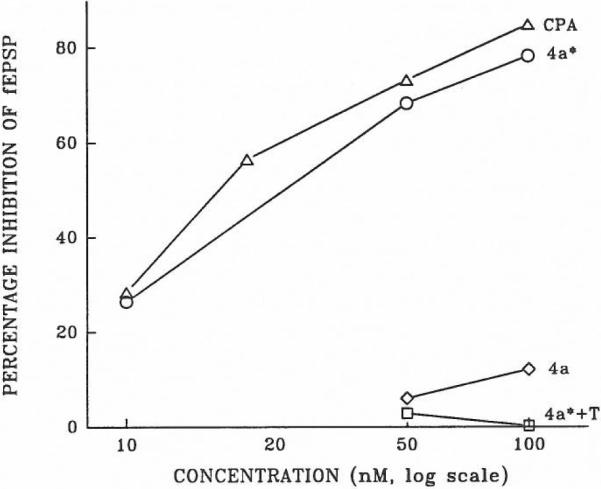
The effect of adenosine derivatives on synaptic potentials in the rat hippocampus. The percentage inhibition of fEPSP responses is shown for CPA, irradiated 4a* (irradiated 4a) and non-irradiated 4a. The effect of irradiated 4a* is also shown when superfused together with 200 μM theophylline (4a* + T). Each point on the graph represents an average of two or three observations.
Discussion
Most of the previously reported “caged” probes were for receptors that govern ion channels directly. The linkage between adenosine receptors and ion channels is indirect, but the outstanding problems in this field warrant the development of “caged” adenosine agonists. Adenosine is a powerful modulator of the biochemical and electrophysiological events in a variety of tissues. These effects are mediated by multiple categories of specific membrane receptors which exhibit restricted regional and cellular distributions. The availability of highly selective agonist and antagonist tigands for the A1-adenosine receptor has facilitated the identification and partial characterization of this receptor subtype. The present study demonstrates that the strategy of utilizing photolabile “caged” receptor agonists for the study of A1-receptors is a viable new approach to further characterize the physiological actions of adenosine.
N6-Cyclopentyiadenosine (CPA) is among the most potent of the A1-selective agonists. CPA inhibits synaptic transmission at nanomolar concentrations, thus it is approximately three orders of magnitude more potent than adenosine itself.22 This neuromodulatory activity can be inhibited by placing a 2-nitrobenzyl group at the S′-position of CPA (compound 4a). The 2-nitrobenzyl group is stable under physiological conditions, superfusion of brain slices with compound 4a for up to one hour did not mimic the inhibitory effect of similar concentrations of CPA. When subjected to photoactivation, compound 4a behaved in a manner similar to native CPA. Photoactivated 4a inhibited synaptic potentials in a dose-dependent manner at nanomolar concentrations, and this effect was blocked by the adenosine receptor antagonist theophylline. Compound 4a thus represents a caged adenosine agonist with potent activity at A1-receptors.
Caged adenosine receptor agonists represent a promising tool for the characterization of the receptors and physiological actions of adenosine. Several applications for these compounds can be anticipated. The rapid kinetics of adenosine′s physiological effects should now be accessible for analysis. One of the main problems associated with the high-affinity A1-receptor agonists is their characteristic limited diffusion into a tissue. For instance, stable responses to CPA require at least 20 min of continuous application in superfused slices of the brain. The slow onset of the inhibitory action of CPA likely reflects its limited penetration through the tissue. Utilizing caged compounds it should be possible to control precisely the biological availability (both timing and concentration) of adenosine agonists. Another potential use of these compounds is the controlled delivery of adenosine agonists to a restricted region of interest. The use of focal irradiation, e.g. with an excimer laser,12 in conjunction with 4a or 4b will allow the presentation of an adenosine agonist to an activated region while transiently maintaining other regions relatively free of irradiated agonist. This might be beneficial in a surgical context in which A1-receptors could be rapidly activated in a restricted region of interest.
Acknowledgements
The authors wish to acknowledge the generous financial support of Cortex Pharmaceuticals, Inc., Irvine CA and NSF grant #RNS-8901154 to K.L., and helpful discussions with Dr. Ray Bartus, Dr. David Eveleth, and Reggie Dean of Cortex. We thank Stephanie Harris for technical assistance.
Footnotes
Dedicated to Prof. A. Patchornik on the occasion of his 65th birthday.
References
- 1.Dunwiddie TV, Proctor W. Mechanisms underlying physiological responses to adenosine in the central nervous system. In: Gerlach E, Becker B, editors. Topics and Perspectives in Adanosine Research. Springer Verlag; 1987. pp. 499–508. [Google Scholar]
- 2.Belardinelli L, Linden J, Berne RM. Prog. Cardiovasc. Dis. 1989;32:73–97. doi: 10.1016/0033-0620(89)90015-7. [DOI] [PubMed] [Google Scholar]
- 3.Churchill PC, Churchill MC. J. Pharmacol. Exp. Thar. 1985;232:589–594. [PubMed] [Google Scholar]
- 4.Bruns RF. Nucleosides and Nucleotides. 1991;10:931–943. [Google Scholar]
- 5.Fredholm BB. Acta Phvsiol. Scand. 1990;140:245–255. doi: 10.1111/j.1748-1716.1990.tb08996.x. [DOI] [PubMed] [Google Scholar]
- 6.Burke SP, Nadler JV. J. Neurochem. 1988;51:1541–1551. doi: 10.1111/j.1471-4159.1988.tb01123.x. [DOI] [PubMed] [Google Scholar]
- 7.Lee KS, Schubert P, Heinemann U. Brain Res. 1984;321:160–164. doi: 10.1016/0006-8993(84)90694-2. [DOI] [PubMed] [Google Scholar]
- 8.Dunwiddie TV, Basile A, Palmer M. Lite Sci. 1984;34:37–47. doi: 10.1016/0024-3205(84)90328-x. [DOI] [PubMed] [Google Scholar]
- 9.Barraco RA. Behavioral Actions of Adenosine and Related Substances. In: Phillis JW, editor. Adenosine and Adenine Nueleotides as Regulators of Cell Function. CRC Press; Boca Raton FL: 1991. pp. 339–366. Chapter 26. [Google Scholar]
- 10.Barraco RA, El-Ridi MR, Ergene E, Phillis JW. Brain Rbs. Bull. 1991;26:59–84. doi: 10.1016/0361-9230(91)90192-m. [DOI] [PubMed] [Google Scholar]
- 11.Gurney AM, Lester HA. Phvsiol. Rev. 1987;67:583–617. doi: 10.1152/physrev.1987.67.2.583. [DOI] [PubMed] [Google Scholar]
- 12.Wilcox M, Viola RW, Johnson KW, Billington AP, Carpenter BK, McCray JA, Guzikowski AP, Hess GP. J. Org. Chem. 1990;55:1585–1589. [Google Scholar]
- 13.a) Patchornik A, Amit B, Woodward RB. J. Amer. Chem. Soc. 1970;92:6333–6335. [Google Scholar]; b) Zehavi U, Amit B, Patchornik A. J. Org. Chem. 1972;37:2281–2284. [Google Scholar]
- 14.Patchornik A, Jacobson KA, Strub M-P. Photo-reversible affinity labeling. In: van Binst G, editor. Design and Synthesis of Organic Molecules Based on Molecular Recognition, Proceedings of the XVIIIth Solvay Conference on Chemistry. Springer; Brussels: 1986. pp. 235–241. [Google Scholar]
- 15.Lee K, Oliver M, Schottler F, Lynch G. Electron microscopic studies of brain slices: the effects of high-frequency stimulation on dentriric ultrastructure. In: Kerkut GA, Wheal HV, editors. Electrophysiologv of Isolated Mammmaiian CNS Preparations. Academic Press; London: 1981. pp. 189–211. [Google Scholar]
- 16.Jacobson KA. Comprehensive Medicinal Chemistry. Vol. 3. Pergamon, Press; London: 1990. Adenosine (P1) and ATP (P2) receptors; pp. 601–642. Chapter 12.10. [Google Scholar]
- 17.Olsson RA, Kusachi S, Thompson RD, Ukena D, Padgett W, Daly JW. J. Med. Chem. 1986;29:1683–1683. doi: 10.1021/jm00159a020. [DOI] [PubMed] [Google Scholar]
- 18.Moos WH, Szotek DS, Bruns RF. J. Med. Chem. 1985;28:1383–1384. doi: 10.1021/jm00148a001. [DOI] [PubMed] [Google Scholar]
- 19.Jacobson KA, Kirk KL, Padgett WL, Daly JW. J. Med. Chem. 1985;28:1341. doi: 10.1021/jm00147a039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchison AJ, Williams M, deJesus R, Oei HH, Ghai GR, Webb RL, Zoganas HC, Stone GA, Jarvis MF. J. Med. Chem. 1990;33:1919–1924. doi: 10.1021/jm00169a015. [DOI] [PubMed] [Google Scholar]
- 21.Ohtsuka E, Tanaka S, Ikehara M. Synthesis. 1977:453–454. [Google Scholar]
- 22.Reddington M, Lee K, Schubert P. Neuroscience Letters. 1982;28:275–279. doi: 10.1016/0304-3940(82)90070-2. [DOI] [PubMed] [Google Scholar]