

 This work is licensed under a
This work is licensed under a Summary
Tumor-induced osteomalacia (TIO) is a rare paraneoplastic syndrome caused primarily by benign mesenchymal tumors. These tumors typically follow a benign clinical course and local recurrence occurs in <5% of cases. We investigated a 49-year-old man with a recurrent mesenchymal phosphaturic tumor showing no signs of malignancy. The patient suffered from chronic muscle weakness, myalgia and cramps. His medical record included the diagnosis of oncogenic osteomalacia, for which he was submitted to tumor resection in the left leg three times before. Laboratory examination showed hypophosphatemia, hyperphosphaturia and an elevated serum FGF23 level. A radical surgical approach (amputation) was advised, however, complete biochemical and clinical remission was not reached. Molecular analysis of the tumor cells demonstrated overexpression of growth factor receptors implicated in tumor angiogenesis and metastatic potential (platelet derived growth factor type A (PDGFRA), PDGFRB and vascular endothelial growth factor receptor) together with increased expression of FGF23, x-linked-phosphate-regulating endopeptidase and KLOTHO. TIO is usually associated with benign phosphauturic tumors and, when identified, resection of the tumor leads to complete remission in the majority of cases. The underlying pathophysiology of recurrences in these tumors is not known. This is the first report showing increased expression of growth factor receptors in a locally aggressive but histopathologically benign phosphaturic mesenchymal tumor.
Learning points
TIO is usually associated with benign soft tissue or bone neoplasms of mesenchymal origin.
These tumors typically follow a benign clinical course and even in the rare malignant cases local recurrence occurs in <5%.
Successful identification and removal of the tumor leads to full recovery in the majority of cases.
Background
Tumor-induced osteomalacia (TIO) is usually associated with benign soft tissue or bone neoplasms of mesenchymal origin and is characterized by excessive renal phosphate leading to hypophosphatemia, inappropriately low-normal levels of 1,25-dihydroxyvitamin D (1,25(OH)2 D) and osteomalacia. Long-term oral supplementation of phosphate and vitamin D may also induce secondary or tertiary hyperparathyroidism, confusing further the clinical picture (1), (2), while co-existence of TIO with primary hyperparathyroidism is rarely seen (3), (4).
These tumors typically follow a benign clinical course and even in the rare malignant cases, local recurrence occurs in <5% and distant metastasis are very uncommon (5).
In this study, we report a case of TIO due to a histopathologically benign phosphaturic mesenchymal tumor behaving in a locally highly invasive and infiltrative manner leading to multiple recurrences. In addition, our patient's case was further complicated with parathyroid hyperplasia that led to significant deterioration of his clinical condition.
Molecular analysis of the tumor cells demonstrated increased expression of growth factor receptors, such as vascular endothelial growth factor receptor and platelet-derived growth factor receptor, implicated in tumor angiogenesis and metastasis of solid tumors.
Case presentation
A 49-year-old male was admitted to the outpatient clinic of our Endocrinology Division complaining of chronic diffuse muscle weakness, myalgia and cramps that were aggravated in the prior 6 months.
The patient's medical record included the diagnosis of oncogenic osteomalacia initially discovered 10 years earlier. In 2004, at the age of 39, he developed diffuse muscle weakness and cramps. His biochemical profile revealed markedly decreased phosphate levels and very low levels of 1,25 (OH)2 vitamin D. The patient was treated with oral phosphate and calcitriol supplementation and showed significant clinical and biochemical improvement. Four years after the initial presentation, the patient started having difficulty walking and developed hemiparesis of the left great toe. Magnetic resonance imaging (MRI) revealed a large mass in the upper part of the left gastrocnemius measuring 10×8×7 cm and infiltrating the upper third of the fibula. Based on the electromyogram results, the patient's paretic symptoms were found to be due to tumor entrapment of the left peroneal nerve, causing dysfunction of the neuromuscular activity. The tumor was then resected and described as a benign phosphaturic mesenchymal tumor without evidence of malignancy in the pathology report. One year later, there was a recurrence of the tumor mass and the patient underwent resection of the remnant tumor. The same year his clinical condition was complicated with the diagnosis of diffuse large B-cell lymphoma (DLBCL), treated with six cycles of R-CHOP with subsequent remission. The patient was systematically treated with oral phosphate and calcitriol and had been relatively stable until further deterioration of his clinical condition occurred.
On admission to our center, the patient had severe hypophosphatemia (1.8 mg/dl, RR: 2.7–4.5 mg/dl) and elevated 24-h urine phosphate (1797.4 mg/24 h, RR: 400–1300 mg/24 h), elevated ALP levels (300 IU/l, RR: 40–150 IU/l), elevated parathyroid hormone (PTH) (19.4 pmol/l, RR: 1.8–6.03 pmol/l) and serum Ca2 + levels (10.8 mg/dl, RR: 8.2–10.6 mg/dl), and low to normal 1,25 (OH)2-vitamin D levels (18 pg/ml, RR:18–24 pg/ml) and 25-OH-vitamin D (20.9 ng/ml, RR: 40–100 ng/ml). The renal threshold phosphate concentration (TmPO4/GFR) as determined by the Walton and Bijvoet nomogram was noted to be low at 0.3 mmol/l (0.8–1.4), confirming the excessive loss of phosphate from the urine. There were no signs of glycosuria, aminoaciduria or proteinuria. MRI located a tumor in the periphery of the head and the upper third of the left fibular diaphysis (Fig. 1). Serum FGF23 levels were as high as 74 000 pg/ml, confirming the diagnosis of the thrice-recurrent phosphaturic tumor. Further surgical evaluation and treatment was advised.
Figure 1.
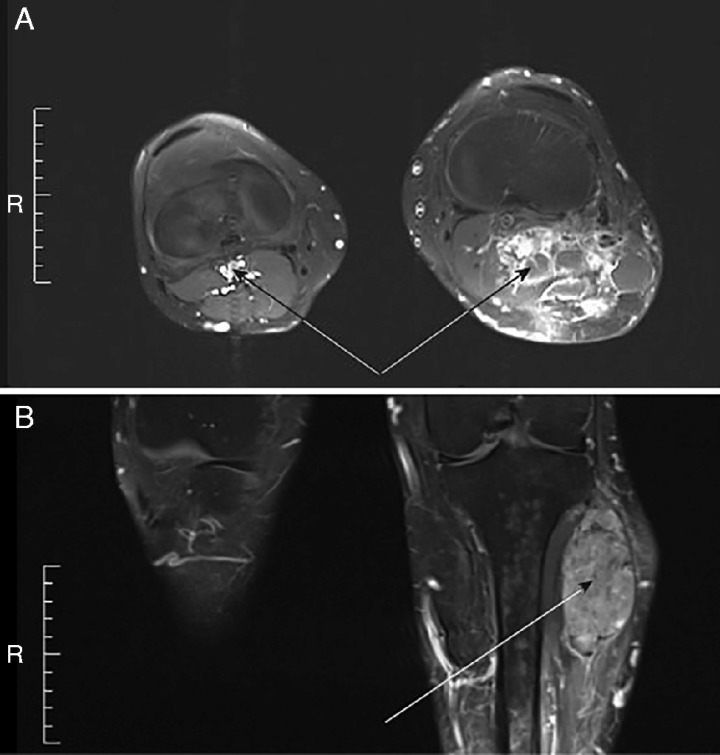
MRI screening of the left calf. (A) Transverse view, (B) Sagittal view. Approximately eight distinct soft tissue lesions around the surgical bed, with abnormal signal and abnormal enhancement, measuring 8 mm to 2.5 cm were detectable. These lesions were located in the periphery of the head and the upper third of the fibular diaphysis and were noted to erode and infiltrate the fibular diaphysis, while infiltrating the adjacent parts of the long extensor muscle of toes and the long peroneal muscle of the outer head of the gastrocnemius muscle (indicated by the arrows). Two of these lesions, measuring 8 and 12 mm, were located in the adjustment parts of the plantaris and posterior tibia muscles respectively.
Investigation
In order to shed more light in the pathophysiology of our patient's phosphaturic tumor, we performed a molecular analysis of samples taken from the tumor and from cells isolated from bone marrow. As controls, we used peripheral blood mononuclear cells (PBMCs) and reference human cancer cell lines. We searched for expression of ERK1, ERK2, mTOR, EGFR, MEK1, MEK2, VEGFR3, AKT1, AKT2, IGFR-1, IGFR-2, PDGFRA, PDGFRB, cMET, FGFR2, FGF23, KLOTHO and x-linked-phosphate-regulating endopeptidase (PHEX) (Table 1).
Table 1.
Sequence of primers. The sequence of primers was run on BLAST to exclude those that would amplify undesired genes
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| 18SrRNA | TGCCCTATCAACTTTCGATGGTAGTC | TTGGATGTGGTAGCCGTTTCTCA |
| ERK1 | GCCCTCCAACCTGCTCATCAA | TACCAGCGCGTAGCCACATACTC |
| ERK2 | AGGCTGTTCCCAAATGCTGACTC | GGCTCGTCACTCGGGTCGTAAT |
| mTOR | CTCACACCCTCCATCCACCTCAT | CCAGGGACGCCAAGACACAGTA |
| EGFR | CCCCTGACTCCGTCCAGTATTGA | CTTTTCCTCCAGAGCCCGACTC |
| MEK1 | TAGTCAACGAGCCTCCTCCAAAACT | TGCTCTCTCTGCGGGGTTTTTTA |
| MEK2 | TGCTGAAAGAGGCCAAGAGGATTC | CGAGGATGTTGGAGGGCTTCAC |
| VEGFR3 | GTGGTCCTTTGGGGTGCTTCTCT | CTCATCCTTGTGCCGTCTCTCAG |
| AKT1 | TTTTGCGGCACACCTGAGTACC | CGACCGCACATCATCTCGTACA |
| AKT2 | ACCGCCCAGTCCATCACAATC | TGCTGGCCGAGTAGGAGAACTG |
| IGFR1 | GCTGGGGAATGGAGTGCTGTATG | CAAACGACCCCTGCCCAAGT |
| IGFR2 | CACCAACCCAAGCACAGGACAC | GGCAGTTTTCATTCTCCCCACAG |
| PDGFRA | CTGAGATGCTTTGGGGAGAGTGAA | TGCTCACTTCCAAGACCGTCACA |
| PDGFRB | CAGACTCCAGGTGTCATCCATCAAC | TTTGCGGGGGTATGTCCACTC |
| cMET | AACAGGTGCAAAGCTGCCAGTG | GCACGCCAAAGGACCACACAT |
| FGFR2 | CTGCGGCCAACACTGTCAAGTT | CCAGTGCTGGTTTCGTACCTTGTAG |
| FGF23 | GGGGCTTTGAGAGACTGGGACTG | CCCTGATTTCTTTCCCCCTGAGT |
| KLOTH | CAGGGACCACCAAGAGAGATGATG | AAGAGTCCACGCCTGATGCTGTA |
| PHEX | CCAGAGCAGAGCATGACATGAAGTC | CGAACTGGGGAATCATAGCACTCA |
Results
None of the tested genes was overexpressed in cells isolated from the bone marrow sample compared to PBMCs from healthy donor or reference cancer cell lines (data not shown). In the tumor sample, we observed an over-expression of VEGFR3, PDGFRA and PDGFRB genes, compared to normal PBMCs (Fig. 2). An over-expression was also observed for PHEX compared to both control samples, while expression of FGF23 was similar between tumor and normal sample, although it was much higher compared to the reference sample (Fig. 2), confirming the diagnosis based on the histopathology reports.
Figure 2.

Gene expression analysis on tissue sample. (A) Relative gene expression of transcription factors in tissue sample, normalized to normal PBMCs. The ΔΔ Ct method was used to perform the analysis. Each bar represents the average of the Ct values. The assays were performed in triplicate and a P value <0.05 was considered significant. Values >0 indicate an increase in gene expression while values <0 indicate a decrease in gene expression. The conditions for subsequent experiments were the same. (B) Relative gene expression of transcription factors in tissue sample, normalized to reference sample (different cancer cell lines). (C) Electrophoresis of qPCR data from tissue sample. The size of the products is mentioned next to each gene. A 100 bp ladder was used as marker. PDGFRA, platelet derived growth factor type A; PDGFRB, platelet-derived growth factor receptor type B, VEGFR3, vascular endothelial growth factor receptor 3; PBMCs, peripheral blood monocytes.
Finally, the KLOTHO gene was expressed in both tumor and marrow samples, but not in control-normal samples, and therefore we were not able to perform a quantitative analysis (Fig. 2).
Treatment
Due to the extension of the lesion and the patient's medical record of repeated recurrences of the tumor, mass amputation of his left limb up to the height of the distal femur was advised.
The histology report revealed multiple regions of an unusual fusocellular neoplasm with elements that suggested a phosphaturic mesenchymal tumor (Fig. 3). There were again no signs of malignancy and the tumor did not appear to be related to the history of the lymphoma.
Figure 3.
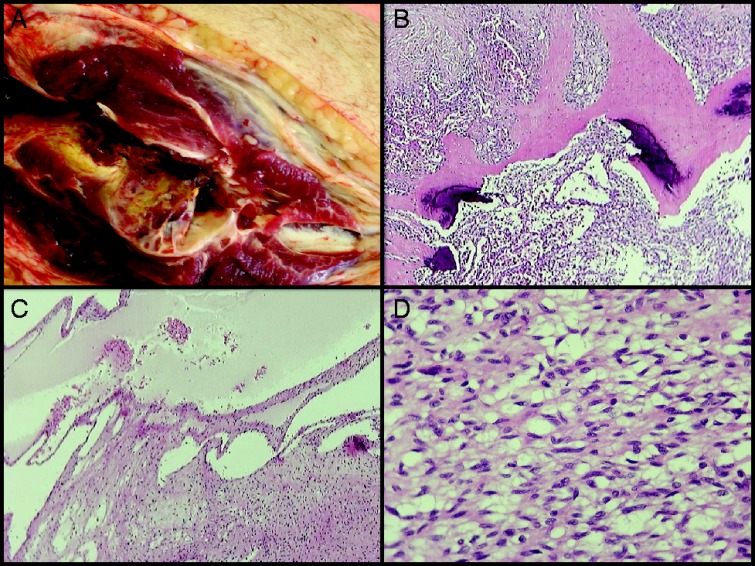
Histology report. Dissection of the specimen showed multiple soft tissue tumors in all muscle compartments of the calf, measuring up to 18 cm (A). A separate lesion was found in the distal tibial metaphysis. Histological examination of all lesions showed a cellular spindle cell neoplasm with variously sized vessels, vascular-like spaces (B) and scattered deposits of calcified extracellular matrix (C). The neoplastic cells were arranged in bundles or diffusely. One small site was found to be highly pleomorphic and with high mitotic index (including some atypical mitosis) was found. The tumor infiltrated skeletal muscles, subcutaneous fat and the proximal end of the fibula. The immunohistochemical examination of the neoplasm showed positivity for vimentin while the markers for SMA, caldesmon, desmin, S-100 protein, EMA and CD34 antigen were negative. The cellular proliferation marker Ki67/MIB1 was positive in <10% of the neoplastic cells. The tibial lesion had identical histology and was separate from all other lesions. There were no signs of DLBCL infiltration. (A) The larger tumor in the upper calf, in close proximity to the proximal end of the fibula infiltrating the adjutant muscle compartments and subcutaneous tissue. (B) H&E stain ×20. Spindle cell neoplasm with both solid pattern and cystic, ‘vascular-like’, spaces. (C) H&E stain ×40. Scattered deposits of calcified extracellular matrix. (D) H&E stain ×400. Cellular spindle cell neoplasm with ‘benign’ histopathological features.
Outcome and follow-up
Two months after amputation, the patient's clinical condition was moderately improved. His laboratory work revealed persistent normal to low PO4 levels (2.1 mg/dl) and elevated, although lower compared to pre-op, FGF23 levels (2630 pg/ml). The patient was discharged supplemented with oral phosphate and calcitriol.
Three months later, due to sustained elevation of serum Ca2 +(11.2 mg/dl) and PTH levels (20 pmol/l), hypercalciuria (361.1 mg/24 h, RR: 100–320 mg/24 h) and phosphaturia (urine PO4=959 mg/24 h, TmPO4/GFR=0.6 mmol/l), the patient submitted to parathyroid scintigraphy that revealed a parathyroid adenoma adjacent to the left inferior pole of the thyroid gland and he had a parathyroidectomy. Histology analysis demonstrated diffuse hyperplasia of the resected parathyroid gland with no signs of malignancy. PTH and serum calcium levels were decreased after parathyroidectomy, but they did not normalize and thus the patient was started on cinacalcet together with oral phosphate and calcitriol.
Whole-body In111 octreotide scintigraphy demonstrated increased uptake in the area of the amputation, which was cautiously attributed to either remaining lesions or activated lymphocytes. Whole-body MRI, however, did not confirm the presence of new lesions. After 3 months of treatment with long-acting octreotide analogue, the patient continues to have low-normal phosphate levels (2.2 mg/dl), increased ALP levels (180 IU/l) and increased FGF23 levels (2730 pg/ml) in the serum. In the patient's last follow-up, we diagnosed a generalized dysfunction of the neuromuscular synapses that aggravated his myopathy due to chronic hypophosphatemia and caused urinary incontinence.
Discussion
Tumors that cause oncogenic osteomalacia are benign mesenchymal tumors, although a great variety of neoplasms have been associated with osteomalacia as a paraneoplasmatic syndrome. In two large series of patients (5), (6), authors have shown that the complete removal of the tumor is curative in the vast majority of cases. Local recurrences are uncommon (6), and in one case of multiple recurrences of a malignant phosphaturic mesenchymal tumor the patient died of his disease 17 years after the first surgery. Our patient's tumor was unusually large and destructive for a phosphaturic mesenchymal tumor, invading all the three compartments of the leg and severely compromising adjacent neuromuscular structure. However, although rare malignant forms have been described before (7), in the histology report of the lesion after the amputation there was still no evidence of malignancy.
The nature of recurrences in these commonly benign phoshpaturic tumors is unknown. In a serial analysis of gene expression in tumors that cause oncogenic osteomalacia, ten genes associated with bone matrix formation, mineral ion transport and bone mineralization were found to be consistently overexpressed in these tumors compared to control-tumors (8). However, none of them has been associated with increased risk of recurrences. Among these FGF23 and secreted frizzled-related protein-4 (sFRP4) genes encode proteins that inhibit phosphate uptake in vitro, while PHEX is usually co-expressed with FGF23 (9). In line with this study we found over-expression of PHEX and FGF23 in our patient's tumor, confirming the laboratory diagnosis of phosphaturic osteomalacia. Interestingly, we also demonstrated expression of KLOTHO in the tumor. KLOTHO is a type 1 transmembrane protein that is implicated in various intracellular signaling and cell-matrix interactions. The formation of KLOTO–FGF receptor complex is necessary in order for FGF23 to activate downstream signaling events (10). To the best of our knowledge, KLOTHO expression has not been reported before in TIO.
Surprisingly, our patients’ tumor overexpressed growth factor receptors that are implicated in cancer growth and metastasis. Platelet-derived growth factors (PDGFs) and vascular endothelial growth factors (VEGFs) activate tyrosine kinase receptors PDGFR (A and B) and VEGFR (1, 2 and 3) respectively (11), (12). Dysregulation of these receptors has been documented in various types of cancers. None of these factors has been reported before in histologically benign phosphaturic mesenchymal tumors. Their overexpression, however, in our patient's peculiar variant could partly explain the extremely infiltrative and invasive behavior of the tumor locally. VEGFR and PDGFR inhibitors would seem a therapeutic potential for our case, but as imatinib, a potent inhibitor of PDGFR, has been associated with severe hypophosphatemia in patients with chronic myeloid leukemia (13), we were reluctant to proceed with this therapeutic approach. FGFR inhibitors could aid in controlling the endocrinologic and metabolic syndrome in this patient, but they are still investigational and unapproved agents. Regorafenib, a potent inhibitor of VEGFR and PDGFR that also inhibits FGFR signaling has been recently approved by FDA for gastrointestinal stromal tumors and metastatic colorectal cancer (14), (15), but since our patient's disease, although locally aggressive and destructive, cannot be defined as malignant, availability of this or similar agents is limited. In addition, none of these agents is anticipated to yield any clinical benefit for the tumor‘s local aggressiveness.
Our patient case was also complicated with parathyroid hyperplasia. The underling pathogenetic mechanisms for secondary or tertiary hyperparathyroidism include postprandial reduction of calcium levels, reduction in calcitriol produced by the renal tubule, and a potential direct stimulatory effect of phosphate on the parathyroid cell itself. Increased FGF23, on the other hand, exerts a negative effect on parathyroid hormone secretion while it increases 1α-hydroxylase activity in the parathyroid cells (3). Previous calcitriol and phosphate treatment in our patient suggests a tertiary rather than primary hyperparathyroidism. The coexistence of hyperparathyroidism with TIO has been described before and can lead to life-threatening hypophosphatemia (4), (5). The use of imaging techniques can help identify the lesion and surgery remains the treatment of choice. In our case, extraction of the affected parathyroid did not lead to the complete remission of the biochemical findings and therefore a calcimimetic was administered to the patient to decrease serum calcium and ameliorate hypophosphatemia.
In conclusion, TIO is a rare and debilitating disease and is associated with both benign and malignant tumors that occur in various locations. Successful identification and removal of the tumor leads to full recovery in the majority of cases. However, multiple recurrences have been described and further research is warranted in order to shed more light in the molecular mechanisms that regulate behavior of these tumors.
Patient consent
This study was approved by the Scientific Review Board of AHEPA University Hospital (protocol number 51741), and is registered on Clinical trials.gov (protocol number NCT01660308). Written informed consent from the patient was obtained before any analysis was performed.
Author contribution statement
N Gerothanasi, A Frydas and E Triantafyllou wrote the first draft. C Poulios and P Hytiroglou performed the histology analysis, P Apostolou and I Papasotiriou performed the molecular analysis, S Tournis performed FGF23 analysis and I Kesisoglou, J G Yovos and M P Yavropoulou revised and approved the final draft.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- 1. Tartaglia F, Minisola S, Sgueglia M, Blasi S, Brunelli D, Degli Effetti E, Maturo A, Cola A, Custureri F & Campana FP. 2006. Tumor-induced hypophosphatemic osteomalacia associated with tertiary hyperparathyroidism: a case report. Il Giornale di Chirurgia 27 9–13. [PubMed] [Google Scholar]
- 2. Tournis S, Samdanis V, Economopoulos D, Giannikou P & Lyritis GP. 2007. Autonomous hyperparathyroidism following long-term phosphate treatment for tumor-induced osteomalacia. Case report and review of the literature. Endocrinologist 17 263–266. 10.1097/TEN.0b013e3181514e2b [DOI] [Google Scholar]
- 3. Elfenbein DM, Weber TJ & Scheri RP. 2012. Tumor-induced osteomalacia masking primary hyperparathyroidism. Surgery 152 1256–1258. 10.1016/j.surg.2012.08.062 [DOI] [PubMed] [Google Scholar]
- 4. Markou A, Tsiama V, Tournis S, Papanastasiou L, Tsiavos V, Dassou A, Vlachou V, Papaliodi E, Asimaki N, Zografos G et al. 2011. Coexistence of tumor-induced osteomalacia and primary hyperparathyroidism. Endocrine Practice 17 e144–e148. 10.4158/EP11177.CR [DOI] [PubMed] [Google Scholar]
- 5. Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, Econs MJ, Inwards CY, Jan de Beur SM & Mentzel T. 2004. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. American Journal of Surgical Pathology 28 1–30. 10.1097/00000478-200401000-00001 [DOI] [PubMed] [Google Scholar]
- 6. Jiang Y, Xia WB, Xing XP, Silva BC, Li M, Wang O, Zhang HB, Li F, Jing HL, Zhong DR et al. 2012. Tumor-induced osteomalacia: an important cause of adult-onset hypophosphatemic osteomalacia in China: report of 39 cases and review of the literature. Journal of Bone and Mineral Research 27 1967–1975. 10.1002/jbmr.1642 [DOI] [PubMed] [Google Scholar]
- 7. Lin HA, Shih SR, Tseng YT, Chen CH, Chiu WY, Hsu CY & Tsai KS. 2014. Ovarian cancer-related hypophosphatemic osteomalacia-a case report. Journal of Clinical Endocrinology and Metabolism 99 4403–4407. 10.1210/jc.2014-2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY et al. 2002. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. Journal of Bone and Mineral Research 17 1102–1110. 10.1359/jbmr.2002.17.6.1102 [DOI] [PubMed] [Google Scholar]
- 9. Bowe AE, Finnegan R, Jan de Beur SM, Cho J, Levine MA, Kumar R & Schiavi SC. 2001. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochemical and Biophysical Research Communications 284 977–981. 10.1006/bbrc.2001.5084 [DOI] [PubMed] [Google Scholar]
- 10. Medici D, Razzaque MS, Deluca S, Rector TL, Hou B, Kang K, Goetz R, Mohammadi M, Kuro OM, Olsen BR et al. 2008. FGF-23-Klotho signaling stimulates proliferation and prevents vitamin D-induced apoptosis. Journal of Cell Biology 182 459–465. 10.1083/jcb.200803024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cebe-Suarez S, Zehnder-Fjallman A & Ballmer-Hofer K. 2006. The role of VEGF receptors in angiogenesis; complex partnerships. Cellular and Molecular Life Sciences 63 601–615. 10.1007/s00018-005-5426-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farooqi AA, Waseem S, Riaz AM, Dilawar BA, Mukhtar S, Minhaj S, Waseem MS, Daniel S, Malik BA, Nawaz A et al. 2011. PDGF: the nuts and bolts of signalling toolbox. Tumour Biology 32 1057–1070. 10.1007/s13277-011-0212-3 [DOI] [PubMed] [Google Scholar]
- 13. Osorio S, Noblejas AG, Duran A & Steegmann JL. 2007. Imatinib mesylate induces hypophosphatemia in patients with chronic myeloid leukemia in late chronic phase, and this effect is associated with response. American Journal of Hematology 82 394–395. 10.1002/ajh.20778 [DOI] [PubMed] [Google Scholar]
- 14. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H et al. 2013. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumors after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381 295–302. 10.1016/S0140-6736(12)61857-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C et al. 2013. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381 303–312. 10.1016/S0140-6736(12)61900-X [DOI] [PubMed] [Google Scholar]