

Abstract
This paper introduces the APPRIS WebServer (http://appris.bioinfo.cnio.es) and WebServices (http://apprisws.bioinfo.cnio.es). Both the web servers and the web services are based around the APPRIS Database, a database that presently houses annotations of splice isoforms for five different vertebrate genomes. The APPRIS WebServer and WebServices provide access to the computational methods implemented in the APPRIS Database, while the APPRIS WebServices also allows retrieval of the annotations. The APPRIS WebServer and WebServices annotate splice isoforms with protein structural and functional features, and with data from cross-species alignments. In addition they can use the annotations of structure, function and conservation to select a single reference isoform for each protein-coding gene (the principal protein isoform). APPRIS principal isoforms have been shown to agree overwhelmingly with the main protein isoform detected in proteomics experiments. The APPRIS WebServer allows for the annotation of splice isoforms for individual genes, and provides a range of visual representations and tools to allow researchers to identify the likely effect of splicing events. The APPRIS WebServices permit users to generate annotations automatically in high throughput mode and to interrogate the annotations in the APPRIS Database. The APPRIS WebServices have been implemented using REST architecture to be flexible, modular and automatic.
INTRODUCTION
The alternative splicing of messenger RNA generates a range of mature RNA transcripts, which if translated into stable proteins, would greatly enrich the repertoire of cellular functions (1,2). The functional annotation of these alternative isoforms presents a serious challenge, not least because of the sheer quantity of genomic data that is being generated by genome annotation projects (3,4). The current human genome GENCODE21 (equivalent to Ensembl 77, (5,6)) currently houses 20 317 protein-coding genes and 93 107 coding transcripts, and the number of annotated transcripts is growing rapidly. Alternative splicing of pre-messenger RNA has been estimated to occur in 95% of multi-exon human genes (7,8).
APPRIS (9) was developed within the GENCODE consortium (5) to address the challenge of annotating alternative protein isoforms with functional information, a topic of growing interest both in normal cells and in disease states (10–12). The APPRIS modules annotate splice isoforms with protein 3D structure information, functionally important residues, Pfam (13) domains, signal peptides and trans-membrane helices, and a score for the cross-species conservation of each transcript model.
What differentiates APPRIS from other methods for annotating splice isoforms is that it uses only the most reliable annotations for protein structure, function and cross-species conservation, and that it uses these annotations to select a single reference CDS as the ‘principal’ isoform. This principal isoform is the one with the most conserved protein features and most evidence of cross-species conservation, while those isoforms with unusual, missing or non-conserved protein features are flagged as alternative (9).
Recent results from our group and others (14,15) suggest that many genes have a single clearly definable dominant protein isoform and that the alternative isoforms are either expressed less frequently, in limited tissues or in unique developmental stages, or have a much shorter half-life.
The standard strategy for determining the dominant protein isoform of a coding gene (16) is to select the longest isoform. For instance databases often choose the longest isoform because it is easier to annotate features to this isoform. However, we have shown that while this method is often correct, it does not have any biological meaning. Between 20 and 25% of the reference isoforms selected by this strategy are likely not to be the main protein product for the gene (9,17) and this number will grow with the expansion of the annotation databases.
In contrast APPRIS principal isoforms coincide with the main isoform detected in proteomics experiments for almost 98% of comparable genes, showing that APPRIS principal isoforms are a clear improvement on choosing the longest isoform as the main protein isoform in the cell (17). The reason that APPRIS is so effective is that most alternative isoforms have either lost regions of conserved structure or function, or have non-conserved exons that are inserted into regions that code for conserved structure or function. The APPRIS Database identifies a principal isoform for 73.33% of human genes annotated in the GENCODE 21 gene set.
The main goal of developing the APPRIS WebServer and WebServices is to allow users to annotate splice isoforms and select a principal isoform for vertebrate genome species beyond those that are annotated in the APPRIS Database, to annotate genes and variants that are missing from the APPRIS Database, and to annotate their experimental results with existing annotations. The APPRIS WebServer has been designed to be used for the comparison of splice isoform annotations for individual genes, while the APPRIS WebServices have been created to allow access to the APPRIS Database and to run an automatic version of the APPRIS server, using REST architecture to be portable, modular and flexible in the automation of programmatic scripts.
METHODS, WEB SERVER AND WEB SERVICES
The APPRIS WebServer and APPRIS WebServices provide annotations for alternative splice isoforms and identify principal isoforms for individual genes. These annotations are based on the modules in the APPRIS Database (9).
The WebServer and WebServices are based on six of the modules of APPRIS Database (see Supplementary Figure S1, see Supplementary ‘APPRIS Modules and Their Scores’). CORSAIR uses BLAST (18) to map (correctly and without gaps) orthologous isoforms in related vertebrate species; CRASH makes conservative predictions of signal peptides using the SignalP and TargetP programs (19); firestar (20) makes reliable predictions of functionally important residues; MATADOR3D checks for the presence of structural homology to 3D structures in the PDB (21); SPADE uses Pfamscan (13) to count conserved and compromised Pfam functional domains; THUMP generates conservative predictions of trans-membrane helices from three separate trans-membrane predictors (22–24).
The principal splice isoform for each gene is selected based on the conservation of protein features, including protein structural and functional data and information from cross-species conservation.
APPRIS WebServer
The APPRIS WebServer can process two types of queries, either the gene name (or Ensembl gene identifier) from specific assembly version, or a set of alternative protein sequences for that gene. In both cases, the species is also required. When the user provides a gene name, the gene is linked to a specific assembly version of Ensembl. At the moment all species, except for human, have a unique assembly version. At present the APPRIS Database houses annotations for five Ensembl species (human, mouse, rat, pig and zebra fish), the APPRIS WebServer allows users to check Ensembl annotations for six other species, dog, cat, cow, opossum, chicken and fugu. If the query gene falls outside these 11 species the user is required to use a set of alternative protein sequences as input.
The report view of APPRIS WebServer displays four sections. The first section shows a panel with information about the query gene. When the query input is protein sequences, the panel contains the identifier of the job and the species name. When the query is a gene name (or Ensembl identifier), the panel contains the name and the identifier of the gene, the species name, the genome location of the gene, and the Ensembl classification (biotype) of the gene. The second section (‘Principal Isoforms’) shows all the variants and highlights the principal isoforms. The isoforms are tagged with the flags PRINCIPAL, and ALTERNATIVE based on the range of protein features. The third section (‘APPRIS annotations’) shows the scores of the APPRIS modules, such as the number of functional residues, the number of whole functional domains, or the vertebrate species conservation score. The APPRIS modules are described in the Supplementary ‘APPRIS Modules and Their Scores’ section.
The last section shows three tab browser panels that allow different views of the annotations. The first browser panel (‘Annotation Browser’) displays the annotations in detail for each isoform. These detailed annotations include information such as the best template of PDB, the best Pfam domain, or the nearest homologue species. The detailed annotations are described in the Supplementary ‘APPRIS Modules and Their Scores’ section. The second browser panel (‘Sequence Browser’) displays the detailed annotations mapped onto the amino acid sequences. The third browser panel (‘Genome Browser’) maps the annotations onto the genomic regions provided by the UCSC Genome Browser (25). The annotations that appear in these browser panels can be filtered by variant and the amino acid sequences of the isoforms can be aligned in the sequence browser panel in ClustalW (26) format. In addition, the APPRIS WebServer supports the downloading and the displaying of data through the website. It should be noted that the UCSC genome browser panel will only be shown for those species where Ensembl and UCSC are using the same build (otherwise the coordinates will be out of phase).
APPRIS WebServices
The APPRIS Database annotations of protein features for human, mouse, rat, pig, and zebra fish splice isoforms are available via web services and the APPRIS WebServices also provides automatic access to the APPRIS server.
APPRIS WebServices make use of standard HTTP method calls (often termed a RESTful services), and then the HTTP request methods GET and POST can be used to send and receive queries and data. The APPRIS WebServices, as APPRIS WebServer, allow the analysis of a specific gene or the sequences of alternative isoforms. These RESTful web services are categorized in ‘runner’ group of services, and have been developed as asynchronous services.
While retrieval services (or some types of analysis) can return a result almost immediately and are suitable for synchronous requests; the request processing of most analyses may be delayed. By calling a web service asynchronously, the client can continue its work without interruption, and will be notified when the asynchronous response is returned. To address these issues we have provided a mechanism for making asynchronous requests: (i) submit a job and get a job identifier (the ‘run’ service), (ii) get the status of a job, an indication of whether the job is pending, running, finished or gave an error (the ‘status’ service), (iii) receive the results of a finished job (the ‘result’ service).
In addition, there are services that retrieve information from specific job analyses and that provide access to the integrated APPRIS Database for the available species. These retrieval services (see Supplementary ‘APPRIS WebServices’ for further details) can be invoked by job identifier, by means of a gene name/identifier, or by means of a genome position. These web services are classified into three broad categories: ‘seeker’, which retrieves information for the available genes or finished job; ‘sequencer’, which retrieves the protein features mapped onto the amino acid sequences; and ‘exporter’, which retrieves information on genes in the APPRIS Database.
While any language capable of making standard HTTP requests can be used, RESTful calls can be accessed using Universal Resource Locators (URLs) by means of a simple web browser query, or from a command-line (using CURL). Client scripts in Perl programming have been provided to allow the execution of APPRIS analyses (‘runner’ RESTful services), and the retrieval of the stored annotations (‘exporter’ RESTful services). The responses of the requests are reported in JSON (by default), GTF, BED or TSV (tabular) format.
System architecture and supported platforms
The APPRIS WebServer (see Figure 1) is designed using the open-source web application framework, AngularJS with back-end servers in Node.js, and Express.js. The interface of RESTful API is created using Swagger, which allows the interaction with the APPRIS WebServices. The software architectural style of REST services has been developed in Perl programming language. The modules involved in the APPRIS analysis are implemented using Perl with required packages, and with the appropriate programs; whose information is stored in an optimized MySQL relational database.
Figure 1.
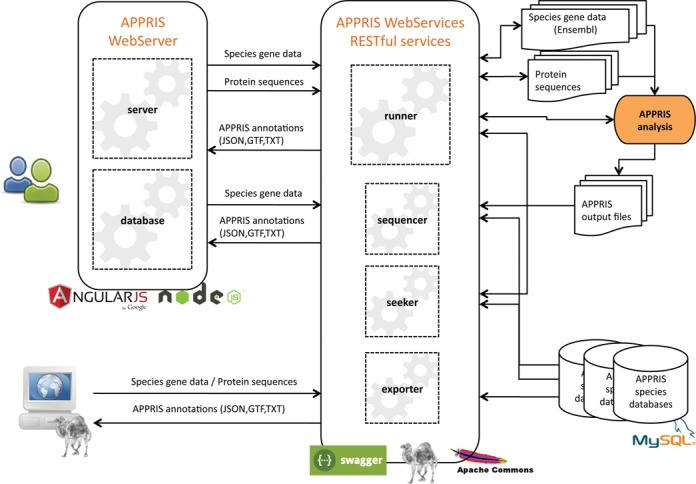
Workflow diagram of APPRIS WebServer and WebServices. The schema represents the organization of server APPRIS WebServer and APPRIS WebServices. The figure also shows the activity of data (inputs/outputs) of the RESTful web services that connect to the web server and to scripts that are capable of making standard HTTP requests. The icons display the tools, frameworks and programming languages used.
APPRIS WebServer has been tested in Mac OS X, Linux and Windows for the browsers Firefox 35.0.1, Google Chrome 40.0.2214.111, and Safari 7.1.3. At this point, it does not support Explorer. Additional support for alternative browsers is in progress.
PRACTICAL CASE
Here, we show one practical example to illustrate the utility of APPRIS WebServer in the selection of principal isoforms (Figure 2). For this example, we use isoforms from the gene ZNF721 (ENSG00000182903) to which we have added a new splice isoform (ZNF721-NEW) to the annotated Ensembl isoforms by combining the ZNF721-009 (ENST00000511833) and ZNF721-002 (ENST00000505900) variants. This new protein sequence was created by adding the translated residues from the first two exons of ENST00000505900 to the translated residues from the third exon of ENST00000511833 (Figure 2A). The APPRIS WebServer is executed submitting the Homo sapiens species name, the set of alternative protein sequences, and the selected methods that will be applied (Figure 2B). A status log panel appears after a submitted a job, indicating whether the job is pending, running, finished or giving an error.
Figure 2.
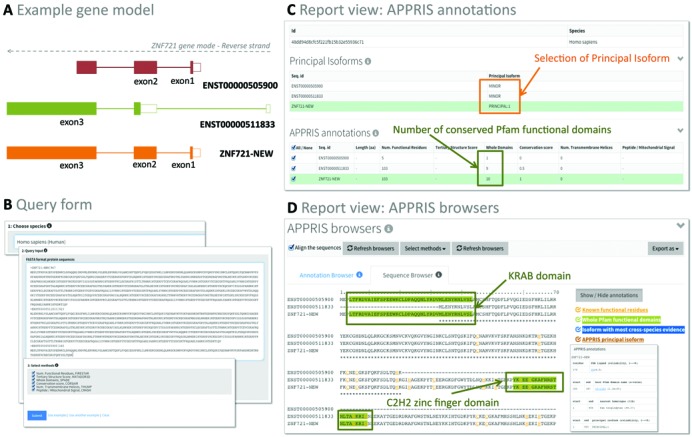
Tutorial Example for APPRIS WebServer (ZNF721). (A) Gene model for ZNF721 showing two Ensembl annotated transcripts, ZNF721-002 (ENST00000505900) and ZNF721-009 (ENST00000511833) and a mock-up of a third transcript, ZNF721-NEW (in orange). The exons from ENST00000505900 and ENST00000511833 that have been used to build the new transcript are labeled. (B) APPRIS WebServer input form showing a query composed by three sequences. Two of them are the protein sequences of ENST00000505900 and ENST00000511833 and the third is the new isoform (ZNF721-NEW) created by joining the first two exons of the ENST00000505900 transcript to the third exon of ENST00000511833. (C) Sections of ‘Principal Isoform’ and ‘APPRIS annotation’ report view. The ZNF721-NEW isoform is selected as the principal isoform, based on the number of Pfam domains. ZNF721-NEW has 10 whole conserved PfamA domains compared to the nine domains in ENST00000511833, and the single domain in ENST00000505900. (D) Snapshot of the ‘Sequence Browser’ panel that shows the annotations mapped onto the alignments of protein sequences. The detailed annotations appear in pop-up windows. The new isoform (ZNF721-NEW) brings together the KRAB domain from ENST00000505900 and the nine C2H2 zinc finger domains from ENST00000511833 (just one highlighted).
The report view of APPRIS annotations (Figure 2C) shows the selection of the new isoform (ZNF721-NEW) as the principal isoform because it has ten whole conserved PfamA domains compared to the nine domains from ENST00000511833, and the single domain in ENST00000505900. The ‘Sequence Browser’ panel (Figure 2D) shows the annotations mapped onto the alignments of sequences. The new isoform (ZNF721-NEW) brings together the Krueppel-associated box (KRAB) domain from ENST00000505900 and the nine C2H2 zinc finger domains from ENST00000511833. KRAB domains are transcription repression modules and are common in C2H2 zinc finger proteins; indeed over 400 human C2H2 zinc finger proteins contain a KRAB domain (27).
DISCUSSION
The APPRIS WebServer and WebServices are tools for the annotation of alternative splice isoforms While the WebServer can be used to annotate individual genes and isoforms with protein structural and functional information and an indication of the cross-species conservation, the WebServices provides access to the existing annotations in the APPRIS Database and allows the automatic use of the annotation modules via the server. APPRIS select a principal isoform for each protein coding gene and the annotations make it possible to predict how alternative splicing events will affect splice isoforms.
We have shown that the principal isoforms selected by APPRIS almost always correspond with the most highly expressed protein isoform, as determined from large-scale proteomics experiments (17). The APPRIS WebServer and WebServices have a wide range of uses, from the determination of principal and alternative isoforms for genes in individual research projects, to the determination of principal and alternative exons for use in genome-wide analysis of variants. The APPRIS Database (9) currently houses splice isoform annotations and principal isoforms for five vertebrate species (human, mouse, rat, pig and zebrafish), and an annotation for Drosophila is close to completion. All these annotations are available through the APPRIS WebServices. APPRIS principal isoforms have been incorporated into the Ensembl annotations (6).
The APPRIS annotations, the WebServer and the WebServices are free, accessible to all and there is no login requirement.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank GENCODE and Ensembl for the dissemination of APPRIS annotations.
FUNDING
National Institutes of Health (NIH) [U41 HG007234]; Spanish National Institute of Bioinformatics (www.inab.org), a platform of the ‘Instituto de Salud Carlos III’ [INB-ISCIII, PRB2 to J.M.R.]. Funding for open access charge: NIH [U41 HG007234].
Conflict of interest statement. None declared.
REFERENCES
- 1.Smith C.W., Valcárcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- 2.Tress M.L., Martelli P.L., Frankish A., Reeves G.A., Wesselink J-J., Yeats C., Olason P.I., Albrecht M., Hegyi H., Giorgetti A., et al. The implications of alternative splicing in the ENCODE protein complement. Proc. Natl, Acad, Sci, U.S.A. 2007;104:5495–5500. doi: 10.1073/pnas.0700800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mudge J.M., Frankish A., Fernandez-Banet J., Alioto T., Derrien T., Howald C., Reymond A., Guigó R., Hubbard T., Harrow J. The origins, evolution, and functional potential of alternative splicing in vertebrates. Mol. Biol. Evol. 2011;28:2949–2959. doi: 10.1093/molbev/msr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frankish A., Mudge J.M., Thomas M., Harrow J. The importance of identifying alternative splicing in vertebrate genome annotation. Database. 2012 doi: 10.1093/database/bas014. doi:10.1093/database/bas014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: the reference annotation for the ENCODE Project. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 8.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez J.M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J.J., Lopez G., Valencia A., Tress M.L. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 2013;41:D110–D117. doi: 10.1093/nar/gks1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.David C.J., Manley J.L. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okumura N., Yoshida H., Kitagishi Y., Nishimura Y., Matsuda S. Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochem. Biophys. Res. Commun. 2011;413:395–399. doi: 10.1016/j.bbrc.2011.08.098. [DOI] [PubMed] [Google Scholar]
- 12.Porola P., Mackiewicz Z., Laine M., Baretto G., Stegaev V., Takakubo Y., Takagi M., Ainola M., Konttinen Y.T. Laminin isoform profiles in salivary glands in Sjögren's syndrome. Adv. Clin. Chem. 2011;55:35–59. doi: 10.1016/b978-0-12-387042-1.00003-4. [DOI] [PubMed] [Google Scholar]
- 13.Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R., Heger A., Hetherington K., Holm L., Mistry J., et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ezkurdia I., Del Pozo A., Frankish A., Rodriguez J.M., Harrow J., Ashman K., Valencia A., Tress M.L. Comparative proteomics reveals a significant bias towards alternative protein isoforms with conserved structure and function. Mol. Biol. Evol. 2012;29:2265–2283. doi: 10.1093/molbev/mss100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheynkman G.M., Shortreed M.R., Frey B.L., Smith L.M. Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Mol. Cell. Proteomics. 2013;12:2341–2353. doi: 10.1074/mcp.O113.028142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boutet E., Lieberherr D., Tognolli M., Schneider M., Bairoch A. UniProtKB/Swiss-Prot. Methods Mol. Biol. 2007;406:89–112. doi: 10.1007/978-1-59745-535-0_4. [DOI] [PubMed] [Google Scholar]
- 17.Ezkurdia I., Rodriguez J.M., Carrillo-de Santa Pau E., Vázquez J., Valencia A., Tress M.L. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res. 2015;14:1880–1887. doi: 10.1021/pr501286b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emanuelsson O., Brunak S., von Heijne G., Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 20.Lopez G., Maietta P., Rodriguez J-M., Valencia A., Tress M.L. firestar–advances in the prediction of functionally important residues. Nucleic Acids Res. 2011;39:W235–W241. doi: 10.1093/nar/gkr437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rose P.W., Beran B., Bi C., Bluhm W.F., Dimitropoulos D., Goodsell D.S., Prlic A., Quesada M., Quinn G.B., Westbrook J.D. The RCSB Protein Data Bank: redesigned web site and web services. Nucleic Acids Res. 2011;39:392–401. doi: 10.1093/nar/gkq1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics. 2007;23:538–544. doi: 10.1093/bioinformatics/btl677. [DOI] [PubMed] [Google Scholar]
- 23.Käll L., Krogh A., Sonnhammer E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Viklund H., Elofsson A. Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci. 2004;13:1908–1917. doi: 10.1110/ps.04625404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–D681. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huntley S., Baggott D.M., Hamilton A.T., Tran-Gyamfi M., Yang S., Kim J., Gordon L., Branscomb E., Stubbs L. A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressor. Genome Res. 2006;16:669–677. doi: 10.1101/gr.4842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.