

Abstract
The Consensus Constrained TOPology prediction (CCTOP; http://cctop.enzim.ttk.mta.hu) server is a web-based application providing transmembrane topology prediction. In addition to utilizing 10 different state-of-the-art topology prediction methods, the CCTOP server incorporates topology information from existing experimental and computational sources available in the PDBTM, TOPDB and TOPDOM databases using the probabilistic framework of hidden Markov model. The server provides the option to precede the topology prediction with signal peptide prediction and transmembrane-globular protein discrimination. The initial result can be recalculated by (de)selecting any of the prediction methods or mapped experiments or by adding user specified constraints. CCTOP showed superior performance to existing approaches. The reliability of each prediction is also calculated, which correlates with the accuracy of the per protein topology prediction. The prediction results and the collected experimental information are visualized on the CCTOP home page and can be downloaded in XML format. Programmable access of the CCTOP server is also available, and an example of client-side script is provided.
INTRODUCTION
Transmembrane proteins (TMPs) play an important role in living cells, both in unicellular and multicellular organisms, since they take part in energy production processes, material and information transports and establish cell-cell adhesions. Previous studies estimate that 20–30% of coded proteins in the various genomes are TMPs (1–3). Despite their abundance and importance, the number of solved TMP structures is relatively low (4–6) compared to those of globular proteins, due to the challenging experimental conditions that these proteins require in structure determination. However, there are several indirect experimental techniques providing information about the localization of TM segments, e.g. experiments based on fusion with reporter enzymes or proteins (7–10), post translational modification (11–14), protease digestion (15), immune-localization (16–20), chemical modification (21–23), etc.
Topology prediction provides a fast, low resolution structural information about TMPs, which could be used as a starting point for laboratory experiments (24) or modeling their 3D structures (25). Most of the early prediction methods were based on the physical-chemical properties of amino acids and were able to determine the topography fairly accurately. Later these approaches were extended with the ‘positive inside’ rule (26), opening prospects to localize the water soluble TMP regions. Machine learning algorithms increased the accuracy of the predictions to a higher level, however these methods depend on the training set used, and therefore their performances are usually lower when encountering new sequence families. As a consequence, whenever a new TM topology is discovered, these methods require re-training (27).
Utilizing experimental evidence (28,29), or combining different algorithms as a consensus predictor could also improve the accuracy by eliminating the errors of individual methods. There are three consensus approaches, two of them TOPCONS (30) and MetaTM (31) are currently available, while ConPredII (32) was not available during the preparation of this work. According to its own benchmarking of TOPCONS, it shows only 1% improvement over single predictors. The per protein accuracy of MetaTM was reported to be 86.3%, which is 4% higher than the best included individual method.
The Consensus Constrained TOPology prediction (CCTOP) method was used to predict the human transmembrane proteome, which is collected in the HTP database (3). For testing the accuracies of the various transmembrane topology predictors on human proteins only, a special benchmark set was compiled, and on this set CCTOP proved to be superior in topology prediction accuracy.
Here we describe the web server of the CCTOP algorithm, a novel consensus topology predictor for α-helical TMPs based on 10 state-of-the-art topology prediction methods. Moreover, the CCTOP server automatically incorporates information from previously determined structural, experimental and bioinformatics studies collected in the PDBTM, TOPDB and TOPDOM databases, respectively. If there are segments with known topology information for the query protein or for any of its homologs in any of the databases mentioned above, this information applied as a constraint in the hidden Markov model. Signal peptide prediction and TMP filtering are also available on the server.
MATERIALS AND METHODS
Preparing benchmark set
A benchmark set was created using the TOPDB database collecting those entries, which have known 3D structures in the PDB that cover the entire TMHs of the TOPDB entry. Redundancy of the sequences was removed at 40% identity using CD-HIT (33), which resulted in 320 sequences. Next, the sequences that were used to train any of the 10 selected methods plus CCTOP itself, were collected, and were used as filters: entries in the testing set with 40% or higher sequence identity with any of the training proteins were removed. This procedure reduced the testing set to 170 sequences, which set was used to measure the accuracy of the different predicting methods.
The CCTOP method
The CCTOP method has three main steps: removing cleavable parts of a target sequence, TMP filtering and topology prediction. Signal peptide segments are often mistaken with TMHs by transmembrane topology prediction algorithms, therefore a preceding analysis of these segments was applied. CCTOP uses SignalP 4.0 (34) to cleave signal peptides; however, this step can be ignored, if a homologous protein in the TOPDB database (35,36) has contradictory evidence.
After removing cleavable segments the next step is to distinguish transmembrane and globular proteins. For this task a simple voting system is applied on the results of Phobius (37), Scampi-single (38) and TMHMM (2,39) algorithms. If any two of these methods predict transmembrane segment(s), the protein is classified as TMP.
A variety of methods was taken into account for the consensus topology prediction, regarding both the training set and the utilized algorithm. Ten methods were selected based on their availability and performance on different benchmark sets: HMMTOP (28,40), MemBrain (41), MEMSAT-SVM (42), Octopus (43), Philius (44), Phobius (37), Pro- and Prodiv-TMHMM (45), Scampi-MSA (38) and TMHMM (2,39). The prediction results of these methods are used as constraints in the same hidden Markov model that was used by HMMTOP but with different weights. The weights depend on the accuracy of each method, measured on a benchmark set collected for the Human Transmembrane Proteome database (3). To further improve the prediction accuracy for each query, its homologous structures from PDBTM (4–6), experiments of homologous sequences from TOPDB (35,36) and conservatively localized domains and motifs from TOPDOM (46) recognized in the query sequence are collected automatically and all these information is incorporated into a probabilistic framework provided by a hidden Markov model as described in Bagos et al. (47). A formalized and more detailed description of the algorithm is available in our earlier paper (3) and on the home page of the CCTOP server.
Calculating the reliability of the prediction
To calculate the reliability of the prediction, posterior probabilities from the HMM are summed up for each main hidden state type (inside, membrane, loop and outside), in each position of the TMP sequence. Reliability is the average of these sums on the most probable state path determined by the Viterbi algorithm (48). The mathematical details can be found on the manual pages of the CCTOP home page.
Evaluating the methods
Prediction accuracies of the 10 selected methods, MetaTM, TOPCONS and CCTOP were tested on the newly compiled benchmark set. For testing, constraints from the PDBTM database were not used, because predefined topology parts would have led to artificially high accuracy values.
The CCTOP server
To handle the high resource consumption of the heavy duty calculations of the different methods, we created a multilayered application architecture. Without going into much detail, the computational part is forwarded to a dedicated load-balanced queue of our HPC cluster, in which we isolated some nodes just to serve these online requests. There are two interface types for using CCTOP: a web server with browser friendly GUI written in C++ using the Wt web toolkit programming library along with our in-house developed XBuilder library developed for our previous works (6,49) and an unpretentious PHP based frontend for scripts. The results can be visually reviewed, and are also available in XML format; its XSD- Schema definition is located at http://cctop.enzim.ttk.mta.hu/CCTOP.xsd.
RESULTS AND DISCUSSION
Evaluation the prediction accuracy of the CCTOP
Previously, the prediction accuracy of the CCTOP algorithm was tested on a benchmark set containing only human proteins (3). To analyze the effect of using a different source of organisms on the prediction accuracy, a new test set was prepared. The 3D structures are known for all proteins in this newly prepared benchmark set, and it does not contain any homologous protein to the training set of any of the 10 used methods nor to the training set of CCTOP.
We have tested the prediction accuracy of various methods on this new benchmark set (Table 1). Some of the algorithms were reported to have 80–90% per protein accuracies; however their performance in this benchmark set are much lower. This can be explained by the stringent filtering of sequences homologous in the training sets. The accuracy of CCTOP was calculated in two ways. In the first test, only the results of the 10 prediction methods were used to calculate the consensus prediction and we did not used any experimental constraints from the PDBTM, TOPDB or TOPDOM databases. TOPCONS was reported to had similar accuracy as the best algorithm utilized, and it was explained as the best input represented the theoretical limit, which cannot be improved (30). However, here we showed with a more sophisticated approach, the prediction accuracy can be significantly improved. In our tests MetaTM shows a setback compared to the incorporated methods. This can be explained with the less strict construction of its benchmark set. Since topology information was not incorporated into the prediction, this test shows that by taking advantage of the probabilistic description of hidden Markov model, the prediction accuracy of a HMM based consensus method can outperform all state-of-the-art and consensus methods.
Table 1. Prediction accuracy (in percent) of the CCTOP compared with the accuracies of the incorporated methods, as well as two consensus methods (TOPCONS and MetaTM) on the newly compiled benchmark set containing 170 proteins.
| CCTOP* | CCTOP | HMMTOP* | MemBrain | MEMSAT-SVM | MetaTM | Octopus | Philius | Phobius | Pro | Prodiv | Scampi-MSA | TMHMM | TOPCONS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sens/res | 98 | 98 | 95 | 92 | 94 | 94 | 93 | 95 | 93 | 96 | 96 | 97 | 93 | 97 |
| Spec/res | 98 | 98 | 95 | 97 | 98 | 97 | 98 | 97 | 97 | 97 | 95 | 97 | 97 | 98 |
| MCC/res | 98 | 98 | 95 | 95 | 96 | 96 | 96 | 96 | 95 | 97 | 96 | 97 | 95 | 97 |
| AccTpg/prot | 84 | 84 | 69 | 62 | 66 | 67 | 71 | 71 | 62 | 75 | 75 | 76 | 66 | 79 |
| AccTop/prot | 81 | 79 | 64 | 0 | 53 | 58 | 66 | 64 | 56 | 70 | 69 | 72 | 59 | 75 |
Predictions marked with * are enhanced with topological constraints from TOPDB and TOPDOM databases. Structural information from PDBTM was not used in any of these tests. Sens/res is the per residues sensitivity, Spec/res is the per residue specificity, MCC/res is the Matthew correlation coefficient, AccTpg/prot is the per protein topography accuracy, AccTpl/prot is the per protein topology accuracy.
In the next test, constraints provided by solved structures were not used, but experiments determining segment localizations relative to the membrane and bioinformatics evidences (conservatively localized domains and motifs) were incorporated. The accuracy of CCTOP increased (Table 1). However, it is important to note that by adding structural constraints the accuracy of CCTOP is expected to be 100% on the benchmark set, as it contains proteins with solved structure.
The reliability of the CCTOP predictions
We define the reliability of the prediction as the average of the posterior probabilities of the main states (inside, outside, membrane, loop) over the best probable state-path (which is the final prediction), which is determined by the Viterbi algorithm. This value can be used to measure the reliability of a single prediction, as it highly correlates with the prediction accuracy. We plotted the per protein prediction accuracy of each subset on the benchmark set which contains predictions above a certain reliability. The independent variable is the coverage, i.e. how many predictions have reliability larger than a certain threshold divided by the number of the proteins in the benchmark set, while the dependent variables are the threshold and the prediction accuracy measured in this subset (Figure 1). As it can be seen, the accuracy values decrease monotonously, showing high correlation with the reliability. In this benchmark set, the accuracy of the predictions with reliability value above 86% is expected to be above 95%.
Figure 1.
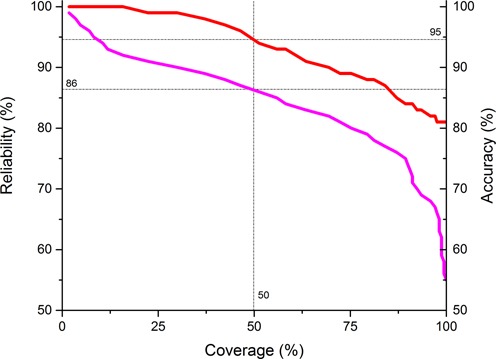
Correlation between the topology accuracy (in percent) of CCTOP and reliability. Predictions are sorted according to their reliability values, and then the topology accuracies and the lowest reliability measured on the subset of the benchmark set (red and magenta, respectively) are plotted against their rank in the sorted list divided by the number of the proteins in the benchmark set (coverage). Above 86% reliability value the prediction accuracy is expected to be 95%.
The home page of the CCTOP server
Protein sequences can be submitted at http://cctop.enzim.ttk.mta.hu. As the prediction time may vary from a few minutes up to 30 minutes depending on sequence length and the load of the various servers incorporated into the CCTOP algorithm, the user may ask an email alert containing a link referring to the results. When the submitted job is finished, a six-panel window is produced by the CCTOP web server.
The first panel is a summary, presenting the most relevant information, protein name (if available), number of predicted TMHs, cross-references to various databases. The generated XML file can be downloaded from the bottom of the panel, or its content can be further investigated in the next panels.
The next panel shows the raw XML file, containing all gathered information together with the results of underlying methods and the final consensus prediction.
The 1D panel shows the amino acid sequence colored by the consensus topology. Colors are based on the localization: gray, black, blue, red, yellow and orange for transit sequence, signal peptide, extra-cytosolic, cytosolic, membrane and re-entrant loop regions, respectively.
The 2D panel is probably the most useful panel. It is a graphical representation of the determined and predicted topology of the given entry. The graph consists of three parts: the final CCTOP prediction, the results of the various topology and topography prediction methods and the collected constraints aligned to the amino acid sequence of the given entry. The x-axis on the graph is the sequence number of the query protein. To inspect the details, the graphs can be enlarged and scrolled. The color code is the same as described above (Figure 2, panel A).
Figure 2.
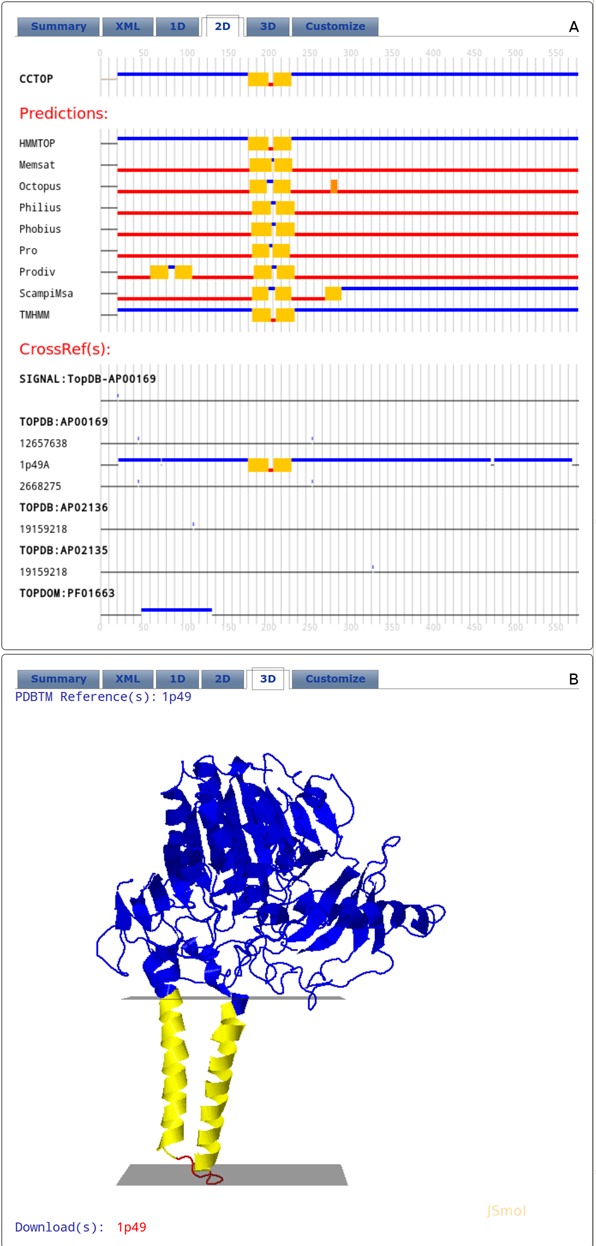
Layout of the result tabs of the CCTOP web server. (A) 2D tab (B) 3D tab. For details, see the text.
If homologous proteins can be found in the PDBTM database, the 3D panel is active and contains their structures. In case of several cross-references to PDBTM each one can be selected and the 3D structure can be inspected using JSMol. Link is provided for each homologous protein to download its 3D co-ordinates from the PDBTM database (Figure 2, panel B).
Finally, using the Customize panel, the initial prediction can be recalculated by (de)selecting any of the prediction methods, mapped experiments or by adding user specified topology constraints. After submission, at the top of this panel, the reliability of the possible localizations for each sequence positions is shown, as well the new reliability value. The content of the other tabs are updated by the results of the recalculated prediction. At the bottom of the page the new XML file can be downloaded, while, the original XML file is still available from the summary panel.
To predict the topology for multiple sequences, a direct interface is available, which allow the programmable access to the CCTOP server. A template python script can be downloaded from the CCTOP server.
The main purpose of the web server is to provide an easy access user interface for the CCTOP method. Some of the utilized methods and CCTOP itself have high computation requirements. Setting up and running these servers locally is rather time consuming. Gathering and mapping already solved structures and experiments together with the prediction results of the state-of-the-art prediction methods are additional non-trivial problems. The CCTOP server does all these tasks automatically.
ENDNOTES
TM: transmembrane; TMP: transmembrane protein; TMH: transmembrane helix.
Acknowledgments
We thank András Fiser and Dániel Kozma for discussion of the manuscript.
FUNDING
Hungarian Scientific Research Fund [K104586; http://www.otka.hu]. ‘Momentum’ Program of the Hungarian Academy of Sciences [to G.E.T.]. Funding for open access charge: Hungarian Scientific Research Fund [K104586; http://www.otka.hu]. ‘Momentum’ Program of the Hungarian Academy of Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wallin E., von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 3.Dobson L., R I., Tusnády G.E. The Human Transmembrane Proteome. Biology Direct. 2015 doi: 10.1186/s13062-015-0061-x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tusnady G.E., Dosztanyi Z., Simon I. Transmembrane proteins in the Protein Data Bank: identification and classification. Bioinformatics. 2004;20:2964–2972. doi: 10.1093/bioinformatics/bth340. [DOI] [PubMed] [Google Scholar]
- 5.Tusnady G.E., Dosztanyi Z., Simon I. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 2005;33:D275–D278. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozma D., Simon I., Tusnady G.E. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013;41:D524–D529. doi: 10.1093/nar/gks1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manoil C., Beckwith J. TnphoA: a transposon probe for protein export signals. Proc. Natl. Acad. Sci. U.S.A. 1985;82:8129–8133. doi: 10.1073/pnas.82.23.8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broome-Smith J.K., Tadayyon M., Zhang Y. Beta-lactamase as a probe of membrane protein assembly and protein export. Mol. Microbiol. 1990;4:1637–1644. doi: 10.1111/j.1365-2958.1990.tb00540.x. [DOI] [PubMed] [Google Scholar]
- 9.Sengstag C., Stirling C., Schekman R., Rine J. Genetic and biochemical evaluation of eucaryotic membrane protein topology: multiple transmembrane domains of Saccharomyces cerevisiae 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mol. Cell. Biol. 1990;10:672–680. doi: 10.1128/mcb.10.2.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brach T., Soyk S., Muller C., Hinz G., Hell R., Brandizzi F., Meyer A.J. Non-invasive topology analysis of membrane proteins in the secretory pathway. Plant J. 2009;57:534–541. doi: 10.1111/j.1365-313X.2008.03704.x. [DOI] [PubMed] [Google Scholar]
- 11.Nilsson I.M., von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 1993;268:5798–5801. [PubMed] [Google Scholar]
- 12.Sokolowska I., Ngounou Wetie A.G., Roy U., Woods A.G., Darie C.C. Mass spectrometry investigation of glycosylation on the NXS/T sites in recombinant glycoproteins. Biochim. Biophys. Acta. 2013;1834:1474–1483. doi: 10.1016/j.bbapap.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 13.Trinidad J.C., Schoepfer R., Burlingame A.L., Medzihradszky K.F. N- and O-glycosylation in the murine synaptosome. Mol. Cell. Proteomic. 2013;12:3474–3488. doi: 10.1074/mcp.M113.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang G., Wu Y., Zhou T., Guo Y., Zheng B., Wang J., Bi Y., Liu F., Zhou Z., Guo X., et al. Mapping of the N-linked glycoproteome of human spermatozoa. J. Proteome Res. 2013;12:5750–5759. doi: 10.1021/pr400753f. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt J.A., Yvone G.M., Brown W.J. Membrane topology of human AGPAT3 (LPAAT3) Biochem. Biophys. Res. Commun. 2010;397:661–667. doi: 10.1016/j.bbrc.2010.05.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charbit A., Ronco J., Michel V., Werts C., Hofnung M. Permissive sites and topology of an outer membrane protein with a reporter epitope. J. Bacteriol. 1991;173:262–275. doi: 10.1128/jb.173.1.262-275.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kast C., Canfield V., Levenson R., Gros P. Transmembrane organization of mouse P-glycoprotein determined by epitope insertion and immunofluorescence. J. Biol. Chem. 1996;271:9240–9248. doi: 10.1074/jbc.271.16.9240. [DOI] [PubMed] [Google Scholar]
- 18.Anand R., Bason L., Saedi M.S., Gerzanich V., Peng X., Lindstrom J. Reporter epitopes: a novel approach to examine transmembrane topology of integral membrane proteins applied to the alpha 1 subunit of the nicotinic acetylcholine receptor. Biochemistry. 1993;32:9975–9984. doi: 10.1021/bi00089a013. [DOI] [PubMed] [Google Scholar]
- 19.Kast C., Gros P. Topology mapping of the amino-terminal half of multidrug resistance-associated protein by epitope insertion and immunofluorescence. J. Biol. Chem. 1997;272:26479–26487. doi: 10.1074/jbc.272.42.26479. [DOI] [PubMed] [Google Scholar]
- 20.Kast C., Gros P. Epitope insertion favors a six transmembrane domain model for the carboxy-terminal portion of the multidrug resistance-associated protein. Biochemistry. 1998;37:2305–2313. doi: 10.1021/bi972332v. [DOI] [PubMed] [Google Scholar]
- 21.Zhu Q., Lee D.W., Casey J.R. Novel topology in C-terminal region of the human plasma membrane anion exchanger, AE1. J. Biol. Chem. 2003;278:3112–3120. doi: 10.1074/jbc.M207797200. [DOI] [PubMed] [Google Scholar]
- 22.Farrell K.B., Tusnady G.E., Eiden M.V. New structural arrangement of the extracellular regions of the phosphate transporter SLC20A1, the receptor for gibbon ape leukemia virus. J. Biol. Chem. 2009;284:29979–29987. doi: 10.1074/jbc.M109.022566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loo T.W., Clarke D.M. Determining the structure and mechanism of the human multidrug resistance P-glycoprotein using cysteine-scanning mutagenesis and thiol-modification techniques. Biochim. Biophys. Acta. 1999;1461:315–325. doi: 10.1016/s0005-2736(99)00165-0. [DOI] [PubMed] [Google Scholar]
- 24.Das S., Hahn Y., Walker D.A., Nagata S., Willingham M.C., Peehl D.M., Bera T.K., Lee B., Pastan I. Topology of NGEP, a prostate-specific cell:cell junction protein widely expressed in many cancers of different grade level. Cancer Res. 2008;68:6306–6312. doi: 10.1158/0008-5472.CAN-08-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H., He Z., Zhang C., Zhang L., Xu D. Transmembrane protein alignment and fold recognition based on predicted topology. PLoS One. 2013;8:e69744. doi: 10.1371/journal.pone.0069744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heijne G. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J. 1986;5:3021–3027. doi: 10.1002/j.1460-2075.1986.tb04601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tusnady G.E., Simon I. Topology prediction of helical transmembrane proteins: how far have we reached. Curr. Protein Peptide Sci. 2010;11:550–561. doi: 10.2174/138920310794109184. [DOI] [PubMed] [Google Scholar]
- 28.Tusnady G.E., Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 29.Bernsel A., Von Heijne G. Improved membrane protein topology prediction by domain assignments. Protein Sci. 2005;14:1723–1728. doi: 10.1110/ps.051395305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernsel A., Viklund H., Hennerdal A., Elofsson A. TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 2009;37:W465–W468. doi: 10.1093/nar/gkp363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klammer M., Messina D.N., Schmitt T., Sonnhammer E.L. MetaTM - a consensus method for transmembrane protein topology prediction. BMC Bioinform. 2009;10:314. doi: 10.1186/1471-2105-10-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arai M., Mitsuke H., Ikeda M., Xia J.X., Kikuchi T., Satake M., Shimizu T. ConPred II: a consensus prediction method for obtaining transmembrane topology models with high reliability. Nucleic Acids Res. 2004;32:W390–W393. doi: 10.1093/nar/gkh380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 34.Petersen T.N., Brunak S., von Heijne G., Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 35.Tusnady G.E., Kalmar L., Simon I. TOPDB: topology data bank of transmembrane proteins. Nucleic Acids Res. 2008;36:D234–D239. doi: 10.1093/nar/gkm751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobson L., Lango T., Remenyi I., Tusnady G.E. Expediting topology data gathering for the TOPDB database. Nucleic Acids Res. 2015;43:D283–D289. doi: 10.1093/nar/gku1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kall L., Krogh A., Sonnhammer E.L. Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 2007;35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernsel A., Viklund H., Falk J., Lindahl E., von Heijne G., Elofsson A. Prediction of membrane-protein topology from first principles. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7177–7181. doi: 10.1073/pnas.0711151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonnhammer E.L., von Heijne G., Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- 40.Tusnady G.E., Simon I. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 1998;283:489–506. doi: 10.1006/jmbi.1998.2107. [DOI] [PubMed] [Google Scholar]
- 41.Shen H., Chou J.J. MemBrain: improving the accuracy of predicting transmembrane helices. PLoS One. 2008;3:e2399. doi: 10.1371/journal.pone.0002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nugent T., Jones D.T. Transmembrane protein topology prediction using support vector machines. BMC Bioinform. 2009;10:159. doi: 10.1186/1471-2105-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viklund H., Elofsson A. OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics. 2008;24:1662–1668. doi: 10.1093/bioinformatics/btn221. [DOI] [PubMed] [Google Scholar]
- 44.Reynolds S.M., Kall L., Riffle M.E., Bilmes J.A., Noble W.S. Transmembrane topology and signal peptide prediction using dynamic bayesian networks. PLoS Comput. Biol. 2008;4:e1000213. doi: 10.1371/journal.pcbi.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viklund H., Elofsson A. Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci. 2004;13:1908–1917. doi: 10.1110/ps.04625404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tusnady G.E., Kalmar L., Hegyi H., Tompa P., Simon I. TOPDOM: database of domains and motifs with conservative location in transmembrane proteins. Bioinformatics. 2008;24:1469–1470. doi: 10.1093/bioinformatics/btn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bagos P.G., Liakopoulos T.D., Hamodrakas S.J. Algorithms for incorporating prior topological information in HMMs: application to transmembrane proteins. BMC Bioinform. 2006;7:189. doi: 10.1186/1471-2105-7-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabiner L.R. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE. 1989;77:257–286. [Google Scholar]
- 49.Kozma D., Simon I., Tusnady G.E. CMWeb: an interactive on-line tool for analysing residue-residue contacts and contact prediction methods. Nucleic Acids Res. 2012;40:W329–W333. doi: 10.1093/nar/gks488. [DOI] [PMC free article] [PubMed] [Google Scholar]