


Abstract
Several algorithms for RNA inverse folding have been used to design synthetic riboswitches, ribozymes and thermoswitches, whose activity has been experimentally validated. The RNAiFold software is unique among approaches for inverse folding in that (exhaustive) constraint programming is used instead of heuristic methods. For that reason, RNAiFold can generate all sequences that fold into the target structure or determine that there is no solution. RNAiFold 2.0 is a complete overhaul of RNAiFold 1.0, rewritten from the now defunct COMET language to C++. The new code properly extends the capabilities of its predecessor by providing a user-friendly pipeline to design synthetic constructs having the functionality of given Rfam families. In addition, the new software supports amino acid constraints, even for proteins translated in different reading frames from overlapping coding sequences; moreover, structure compatibility/incompatibility constraints have been expanded. With these features, RNAiFold 2.0 allows the user to design single RNA molecules as well as hybridization complexes of two RNA molecules. Availability: the web server, source code and linux binaries are publicly accessible at http://bioinformatics.bc.edu/clotelab/RNAiFold2.0.
INTRODUCTION
RNA inverse folding is the problem to determine one (or all) RNA sequences, whose minimum free energy (MFE) secondary structure is identical to a given target secondary structure. Most algorithms for inverse folding use heuristics, such as ensemble defect optimization (1), genetic algorithms (2–4), simulated annealing (5), initial sequence optimization (6), adaptive walk (7,8), etc. Some algorithms, such as NUPACK-Design, do not attempt to solve the inverse folding problem, but instead minimize ensemble defect, which measures the extent to which low energy structures deviate from the target structure. In contrast to other approaches, RNAiFold (9) is the only exhaustive, nonheuristic method, achieved by Constraint Programming (CP). RNAiFold has been used to computationally design functional, synthetic ribozymes, whereby cleavage kinetics have been experimentally determined (10) and to detect novel IRES-like (internal ribosomal entry site) elements validated by a luciferase reporter assay (11). In this note, we describe differences between RNAiFold 1.0 (12) and RNAiFold 2.0.
First, RNAiFold 2.0 is a complete overhaul and reimplementation of all the algorithms from (9) in C++ using the new OR-Tools engine https://code.google.com/p/or-tools/. Since the COMET engine for RNAiFold 1.0 is now obsolete, and no new licenses will be issued, it is not possible for users to execute our COMET source code. In contrast, our new code is now available along with the publicly available engine OR-Tools, supported by Google. Both can be installed and executed by users on various Operating Systems with a C++ compiler. Second, RNAiFold 2.0 allows the user to require solutions to be compatible with a second given structure, in addition to folding into the target structure, and/or be incompatible with base pair formation at positions listed in a prohibition list. Moreover, amino acid constraints have been added, requiring solutions not only to fold into a target structure, but also to code a given protein (or to code for the most similar protein, as determined by the BLOSUM62 similarity matrix). In addition, the user can choose to use Turner’99 [resp. Turner’04] energy parameters (13) by interfacing with Vienna RNA Package 1.8.5 or 2.1.7 (14). Third, RNAiFold 2.0 provides a distinct, novel web service for a fully automated pipeline to design synthetic RNAs, such as the synthetic hammerheads described in (10). In this case, the user can specify a family from Rfam 12.0 (15), then select a member of the automatically generated list of Rfam seed sequences whose MFE structure coincides with the (functional) Rfam consensus structure and then set a threshold for sequence conservation. RNAiFold 2.0 then computes a list of synthetic RNAs, which fold into the (functional) target Rfam consensus structure, and are guaranteed to contain those presumably important nucleotides located at positions which exceed the user-specified sequence conservation threshold.
The plan of this paper is to discuss (i) the automated synthetic design pipeline, (ii) amino acid sequence and prohibited base pair constraints—all of which are not present in our earlier software RNAiFold 1.0 (12)—and to present (iii) a comparison of RNAiFold 2.0, RNAiFold 1.0 and other inverse folding software.
RFAM-BASE DESIGN PIPELINE
Details of the novel method for synthetic RNA design are described in Dotu et al., (10), which additionally discusses the selection criteria (pointwise entropy, ensemble defect, etc.) used to prioritize synthetic type III hammerhead ribozyme candidates for experimental validation. The Rfam-based design pipeline is now an integral part of the RNAiFold 2.0 web server, so we describe here how to fill in the web pages displayed in Figures 1 and 2, in order to design synthetic RNAs likely to function similarly to RNAs in a user-specified family from Rfam 12.0 (15). See the online manual for more information.
Figure 1.
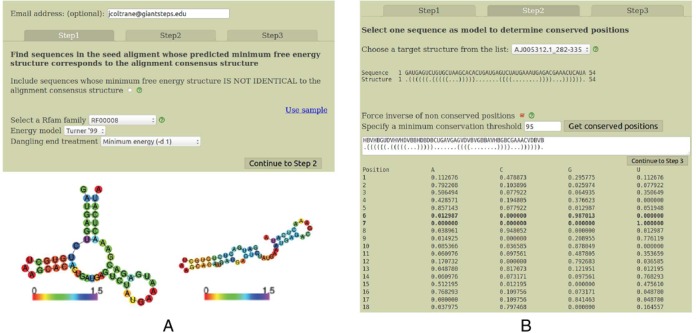
(A) First of three screens in RNA Rfam-based Design, invoked to automate the generation of synthetic RNAs likely to adopt the same function as RNAs in a user-specified Rfam class. Computations may take long, so though optional, it is advisable to enter an email address to be informed of the results when ready. The user must select an Rfam family and the energy model, i.e. Turner’99 or Turner’04 (13) together with a dangle state. As shown in the figure, the Turner’99 parameters (Vienna 1.8.5) can prove to be a better choice than the Turner’04 parameters (Vienna 2.1.7) in certain circumstances—here, Vienna 1.8.5 predicts the correct, functional structure for the hammerhead type III ribozyme (left image) from Peach Latent Mosaic Viroid (PLMVd) with accession code AJ005312.1/282-335, while the Vienna 2.1.7 predicts the incorrect structure (right image). (B) Second of three screens in RNA Rfam-based Design, where the user selects a sequence in the pull-down menu; this sequence, which belongs to the chosen Rfam family will serve as an initial model to generate synthetic sequences. Each displayed sequence folds into the Rfam consensus structure when using the selected energy parameters (if no sequence is shown, then no sequence has this property). In this screen, the user may specify that RNAiFold 2.0 automatically generate sequence constraints for positions that are conserved in the Rfam seed alignment to user-specified minimum threshold; to avoid generating solutions that are too similar to the model sequence, the server automatically generates IUPAC constraints to disagree with the model sequence at all positions where the seed alignment has less than the specified conservation threshold. The position-specific compositional frequency (profile) of the Rfam seed alignment is displayed for each position.
Figure 2.
(A) Third of three screens in Rfam-based Design, where the user can enter additional structure compatibility and incompatibility constraints, which require all solutions to be compatible with a second structure (in addition to folding into the target structure) and which do not allow base pairing at positions stipulated in the incompatibility constraints. (B) Output from the pipeline described in the three previous screen shots. Note that the GC-content, average positional entropy, ensemble defect and other structural diversity measures are computed. These measures provide an idea of how similar the low energy ensemble of structures resembles the minimum free energy structure, which is guaranteed to be identical to the user-input target structure.
Step 1, depicted in the left panel of Figure 1
Though not required, it is useful to enter an email address for notification when the computation terminated in the case of a long job. First, the user should decide whether to check the checkbox that subsequently allows the the selection of sequences whose MFE structure is perhaps not identical to the Rfam consensus structure (explained below). Next, select an Rfam family—in the case of hammerhead III ribozymes, this is RF00008. Next, select the energy model—either Turner’99 or Turner’04, for which energy parameters are described in (13). It is commonly held that Turner’04 parameters are more accurate, though this is not necessarily the case, since Vienna RNA Package RNAfold (14) predicts the correct, functional structure for Peach Latent Mosaic Viroid (PLMVd) hammerhead ribozyme AJ005312.1/282-335 using the Turner’99 parameters (left panel of Figure 1, left image), while the incorrect structure is predicted using the Turner’04 parameters (left panel of Figure 1, right image). Choose the treatment of dangles (stacked, single-stranded nucleotides), where choices are no dangle (-d 0), the minimum of 5′- and 3′-dangle (-d 1), the sum of 5′- and 3′-dangle (-d 2), minimum of 5′- and 3′-dangle plus coaxial stacking (-d 3). For design of functional hammerheads in (10), we used the Turner’99 model with (-d 1), since the MFE structure of PLMVd AJ005312.1/282-335 is identical to the Rfam consensus structure.
Step 2, depicted in the right panel of Figure 1
A pull-down menu allows one to select a target structure from those Rfam seed alignment sequences, whose MFE structure is identical (or similar) to the corresponding Rfam consensus structure. The Rfam consensus structure for a given sequence is determined by placing base pairs in positions dictated by the Rfam consensus (indicated by angle brackets at the bottom of alignments in Stockholm format), then removing base pairs if the nucleotides do not form a Watson–Crick or wobble pair and finally removing base pairs at positions i, j when i < j < i + 4. If the user did not check the checkbox which allows consideration of sequences, whose MFE structure is not identical with the Rfam consensus structure, then it can happen that no target structure will be displayed—indeed, this will happen if no Rfam sequences fold using Turner parameters into their corresponding consensus structure. If the checkbox was checked in Step 1, then sequences whose MFE structure closely resembles the Rfam consensus structure will be displayed and the base pair distance between the consensus and MFE structure will be indicated in parenthesis. After selecting a target structure, the user should set a conservation threshold θ, whose default value is 95%. The server determines the compositional frequency as each position of the selected structure and sets a sequence constraint for those positions whose compositional frequency exceeds threshold θ. The user may check the box which additionally sets a sequence constraint for all remaining positions to be different from the nucleotide of the Rfam sequence whose target structure has been selected—for instance, the largest nucleotide frequency at position 1 of the Rfam alignment is P(C) = 0.478873, which is less than the conservation threshold of 0.95, and since Rfam sequence AJ005312.1/282-335 contains G at position 1, the sequence constraint contains IUPAC code H (not G) at position 1. The resulting target structure and sequence constraint is then displayed.
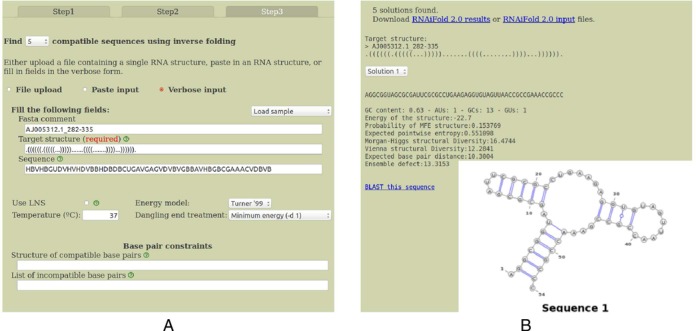
Step 3, depicted in the left panel of Figure 2
By clicking on the button Continue to Step 3, located in the bottom right of Step 2, the FASTA comment, target structure, sequence constraints, energy model, dangle treatment, etc. are automatically entered in the appropriate places in the form in Step 3. The user can choose to generate 1, 5, 10, 50 or MAX (maximum number of solutions that can be computed within a system-dependent resource bound on computation time). Additional constraints can be added at this point. For instance, the user could require all solutions be compatible with the additional structure .............(((((.......)))))............... for which RNAiFold 2.0 correctly returns the solution AGGCGGUAAC CCGAUCCGGG UCUGAAGAGC UCGAGUUAAA GGGCGAAACC GCCC.

Note that (exhaustive) CP is used, rather than (heuristic) Large Neighborhood Search (LNS), as indicated by 0 following #LNS—this is the default, unless otherwise indicated. Additional constraints can be included in the input file, by using the appropriate label preceded by the ‘pound’ symbol (‘#’), where the desired value appears in the next line (see online manual for more details). When running a local copy of the executable, one uses command-line flags, as in RNAiFold2_2.1.7 -RNAscdstr ‘(((...)))’ -RNAseqcon NNNAAANNN.
The right panel of Figure 2 shows the output of the RNAiFold 2.0 web server when designing hammerhead ribozymes—cf. ‘Use sample’ link visible in left panel of Figure 1. A pull-down menu displays each of the solutions found within the system-dependent time limit. For each solution, the secondary structure is displayed (identical to the target structure) and as well as summary information for GC-content, Boltzmann probability of target MFE structure, average pointwise entropy (also called positional entropy) (16), Morgan-Higgs and Vienna structural Diversity (17), expected base pair distance from target (9) and ensemble defect (18). These measures can be used to prioritize the selection of candidates for experimental validation (10). Finally, a link to NCBI BLAST is provided to search for sequences similar to the RNAiFold 2.0 solution sequences.
NOVEL FEATURES IN RNAIFOLD 2.0
Apart from the synthetic RNA design pipeline described in the previous section, RNAiFold 2.0 provides a number of novel features not available in RNAiFold 1.0: (i) the user can choose to use either the Turner99 or Turner04 energy parameters by the built-in interface with Vienna RNA Package 1.8.5 or 2.1.7; (ii) the target can be specified using expanded dot-bracket notation, where a comma indicates that the position may be paired or not; i.e. RNAiFold 2.0 now supports partial targets; (iii) structural constraints have been expanded—in addition to folding into the target structure, solutions can be required to be compatible with an additional structure and can be required to be incompatible with base pair formation at those positions listed in a prohibition list; (iv) amino acid constraints have been added, which require solutions not only to fold into a target structure, but also to code a given protein (or to code for the most similar protein, as determined by the BLOSUM62 similarity matrix); (v) the flag RandomAssignment can be set, which randomizes instantiation order of variable values (used to provide a more unbiased sample of solutions when search space is very large).
Base pair formation may be prohibited using three different syntaxes. (i) If a secondary structure ‘s’ is listed after the flag IncompBP, then positions (i, j) where a base pair occurs in s are not allowed to pair in every solution returned. (ii) The syntax P i j k may be used, which prevents position i from pairing with j, j + 1, j + 2, …, j + (k − 1). (iii) A comma separated list of pairs i1j1,..., injn can be specified, which prevents position i1 from pairing with j1, position i2 from pairing with j2, etc. The user may combine syntax from (ii) and (iii) together, as shown below. Since structural compatibility constraints were illustrated in the previous section, we illustrate the use of constraints (a)-(d) without again demonstrating structural compatibility constraints.
The user first selects the energy model (Turner’99 or Turner’04) from the web page—here, Turner’04 was selected. Additional parameters can be set within the web page form, or within a command file that is uploaded, and shown below. Defaults are taken, unless otherwise mentioned in the command file or web page. Consider the following example, where a stem-loop partial structure is given on the left, with A at position 1, a GNRA-tetraloop at positions 6-9 and a partial structure consisting of a base pair (17, 26) as indicated on the right fragment of the structure. Commas appear at position 16, 18–25, 27, to specify that these positions may be paired or unpaired. Additionally, the first position is prohibited from pairing with any other nucleotide in this 27-nt sequence (P 1 2 26); position 3 is prohibited from pairing with positions 16 and 17 (P 3 16 2), and the nucleotides at positions 4,17 and 4,18 and 4,19 are prohibited from pairing.
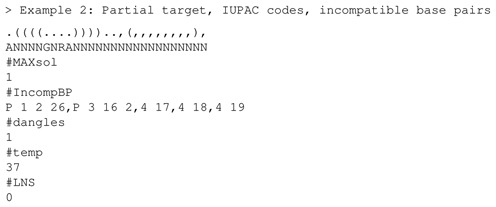
The solution returned in 2.18 s is the following:

The first structure in the output is the user-specified partial target. The solution is given in the third line, followed by GC-content, number of base pairs of each type and the MFE structure of the solution. As required, the MFE structure of each solution agrees with the target (partial) structure at positions occupied by a dot, left parenthesis or right parenthesis, but may differ in positions corresponding to commas in the target (partial) structure. No U's occur in positions 2–27, as required by the prohibition P 1 2 26, nor can 3 form a base pair with 16,17, nor can 4 form a base pair with 17,18,19. Finally, note that the command file may be uploaded or its contents may be copied into the web form text area when the option ‘paste input’ is selected. This may save time, if many options and parameters need to be given.
Finally, amino acid constraints may be specified by using the flag AAseqcon, followed by one or more amino acid sequences, followed by the flag AAstartPos after which the starting position of the first codon of each amino acid sequence is given. Note that there is no bound on the number of (possibly overlapping) coding regions for distinct peptides. In the following example, the target structure has length 52, positions 1–51 code for the peptide FFREDLAFPQGKAREFS and positions 2–52 code for the peptide FLGKIWPSHKGRPGNFL. The flag AAsimilCstr specifies whether the solution must exactly code the given peptides (value 5) or whether each amino acid coded by the returned solutions must have BLOSUM62 similarity at least x (for value x < 5) with each amino acid of the given peptide. Values 6,7 allow the user to enter specific symbols that designate chemical properties of residues coded by all solutions returned—e.g. hydrophobic, positively or negatively charged, polar, etc. (see online manual for details).

Note that flag MAXsol has the value 0, which allows the user to run RNAiFold 2.0 locally with no upper bound on the number of solutions returned. In this case, RNAiFold 2.0 will either terminate with all possible solutions, or the process will die after memory exhaustion, or the user can terminate the process; however, in all cases the output can be saved to a file.
Another novel feature of amino acid constraints is that the flag MaxBlosumScore allows RNAiFold 2.0 to determine a solution of inverse folding for which the BLOSUM62 similarity to the target peptide is an absolute maximum; i.e. no other solution of inverse folding codes a peptide having larger BLOSUM62 similarity to the specified target peptide.
COMPARISON WITH OTHER SOFTWARE
Tables 1, 2 and Figure 3 provide a comparison of all current inverse folding software and web servers. Table 1 gives an overview of distinctive features of each software, while Table 2 gives an overview of the quality and quantity of solution sequences returned by each method.
Table 1. Comparison table for RNA inverse folding software.
| Software | ⇓ | WS | PK | H | MT | PT | T | EM | D | SeqC | StrC | AaC | O | Num |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNAiFold 2.0 | ✓ | ✓ | ✓ | ✓ | ✓ | ’99,’04 | 0,1,2,3 | ✓ | ✓ | ✓ | mfe | MAX | ||
| RNAinverse | ✓ | ✓ | ✓ | ’99,’04 | 0,1,2,3 | IUPAC⋆ | mfe, prob | 100 | ||||||
| RNA-SSD | ✓ | ✓ | ’99 | 1 | IUPAC⋆ | mfe | 10 | |||||||
| Info-RNA | ✓ | ✓ | ’04 | 1 | IUPAC | mfe, prob | 50 | |||||||
| NUPACK | ✓ | ✓ | ✓⋆ | ✓ | ’99,’04 | 0,1,2 | ✓ | ens def | 10 | |||||
| MODENA | ✓ | ✓ | I | def | mfe, prob | ? | ||||||||
| Frnakenstein | ✓ | ✓ | ✓ | I | def | various | ? | |||||||
| IncaRNAtion | ✓ | ✓ | ’04⋆ | IUPAC | pf sampling | |||||||||
| ERD | ✓ | ✓ | ✓ | I | def | IUPAC⋆ | mfe | MAX⋆ | ||||||
| RNAdesign | ✓ | ✓ | ✓ | ’04 | def | various | ||||||||
| RNAfbinv | ✓ | ✓ | ’99, I | def | local A,C,G,U | mfe |
Column headers: Soft (Software), ⇓ (software can be downloaded), WS (web server), PK (pseudoknots), H (hybridization), MT (multiple targets), PT (partial targets), T (temperature), EM (energy model), D (dangles), SeqC (sequence constraints), StrC (structural constraints), AaC (amino acid constraints), O (objective), Num (maximum number of sequences returned). Comments: in column H, RNAiFold 2.0 and NUPACK are the only programs that solve inverse folding for target hybridizations; moreover, NUPACK has ‘✓⋆’, since it is the only algorithm that allows hybridization of more than 2 strands. In column EM, values are ′99 (Turner′99), ′04 (Turner′04), ′04⋆ (Turner′04 base stacking parameters with no entropic free energies), I (installed, depending on the version of Vienna RNA Package installed on user's computer). In column D, dangle status is 0 (no dangle), 1 (max of 5′ and 3′-dangle), 2 (sum of 5′ and 3′-dangle), 3 (dangles and coaxial stacking), def (depending on default setting of user's version of Vienna RNA Package). In column SeqC, values are ✓ (IUPAC plus additional constraints) IUPAC, IUPAC⋆ (limited subset of IUPAC symbols) and local A,C,G,U (oligonucleotide specified at a given position using only A,C,G,U). In column O, values are mfe (minimum free energy structure), prob (maximize Boltzmann probability), ens def (ensemble defect), pf sampling (partition function sampling with a restriction of Turner’04). In column Num, the number of solutions returned by the web server is given (—if no web server available); a question mark in this column appears for MODENA and Frnakenstein, which are genetic algorithms and have a population of evolving sequences, so the user cannot request a fixed number of solutions. ERD contains MAX⋆, since the web server allows the user to request an arbitrary number of iterations (distinct runs) of the program, where 10 min is the maximum computation time allotted per request. In contrast, RNAiFold 2.0 contains MAX in this column, which indicates that as many solutions are returned as possible within the system-dependent run time bound.
Table 2. Comparison of 10 programs for RNA inverse folding, benchmarked on 63 target structures, as explained in the text.
| Method | ERD | FRNA | Incarnation | Info-RNA | MODENA | Nupack | RNA-SSD | RNAfbinv | RNAiFold2 | RNAinverse |
|---|---|---|---|---|---|---|---|---|---|---|
| Output (%) | 100% | 30% | 60% | 95% | 60% | 57% | 90% | 13% | 65% | 65% |
| Target (%) | 85% | 38% | 0% | 57% | 45% | 70% | 82% | 0% | 100% | 18% |
| Avg str len | 397 | 122 | 352 | 393 | 234 | 256 | 400 | 74 | 363 | 208 |
| Avg output | 117 | 325 | 41 535 | 195 | 50 | 22 | 1 | 2 | 55,476 | 935 |
| P(S) | 3.32% | 1.70% | 0.06% | 3.17% | 11.30% | 30.01% | 2.24% | 0.36% | 23.21% | 0.78% |
| Native cont. (%) | 85 ± 9 | 61 ± 15 | 63 ± 13 | 76 ± 12 | 89 ± 9 | 98 ± 1 | 85 | 32 ± 6 | 93 ± 2 | 57 ± 12 |
| Avg E | −0.41 | −0.24 | −0.46 | −0.63 | −0.46 | −0.44 | −0.30 | −0.14 | −0.56 | −0.23 |
| Pos entropy | 0.33 | 0.71 | 0.41 | 0.44 | 0.15 | 0.07 | 0.36 | 0.88 | 0.12 | 0.80 |
| MH diversity | 0.16 | 0.35 | 0.21 | 0.22 | 0.07 | 0.03 | 0.18 | 0.45 | 0.06 | 0.38 |
| Vienna diversity | 0.11 | 0.23 | 0.15 | 0.16 | 0.05 | 0.02 | 0.11 | 0.30 | 0.05 | 0.26 |
| Exp bp dist | 0.09 | 0.21 | 0.27 | 0.16 | 0.06 | 0.01 | 0.08 | 0.38 | 0.03 | 0.24 |
| Ens def | 0.14 | 0.32 | 0.39 | 0.22 | 0.08 | 0.02 | 0.14 | 0.56 | 0.04 | 0.37 |
| Exp num bp | 0.28 | 0.29 | 0.34 | 0.30 | 0.26 | 0.29 | 0.28 | 0.28 | 0.27 | 0.28 |
| GC-content (%) | 55% | 49% | 71% | 72% | 50% | 57% | 36% | 51% | 57% | 49% |
Averages are given, rounded either to two decimals or to the nearest integer as appropriate. Complete data, with averages and standard deviations, can be found on the web server RNAiFold 2.0. FRNA stands for FRNAnkenstein. Row labels are as follows, whereby measures appearing after the double line have been normalized by dividing by sequence length—for instance, Avg E denotes the normalized average free energy of the returned sequences, computed as the average, taken over all 63 individual target structures S0, of average normalized free energies E(a, S0)/|a|, taken over all sequences a returned for target structure S0, where E(a, S0) denotes the free energy of sequence a with respect to the structure S0. The other normalized measures are defined in an analogous manner. (Unnormalized measures) Output (%): fraction of the 63 target structures for which some output was produced. Target (%): average fraction of output sequences whose MFE structure is the target. Avg str len: average target structure length, taken over those target structures for which at least one output sequence was returned. Avg output: total number of sequences returned for all 63 targets, divided by the number of targets for which at least one sequence was returned. P(S): average probability of target structure, defined as the average, taken over all 63 target structures S0, of the average Boltzmann probability P(s, S0) = (exp ( −E(s, S0)/RT)/Z, taken over all sequences s returned for target structure S0. (Normalized measures) Avg E: normalized average free energy with respect to target (previously defined). The remain measures are length-normalized versions of positional entropy, Morgan-Higgs diversity, Vienna diversity, expected base pair distance from target structure, ensemble defect with respect to target structure, expected number of base pairs, proportion of native contacts and GC-content. Measures are defined in the text.
Figure 3.
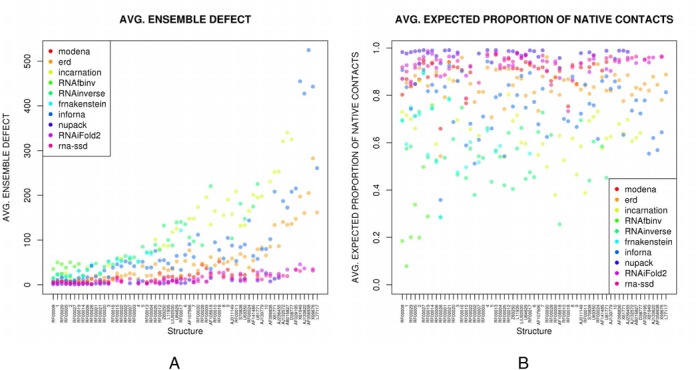
(A) Average ensemble defect for inverse folding software, where the (non length-normalized) averages are taken over all sequences returned for a fixed target structure. Name of the target structure is given on the x-axis, arranged in increasing length (length is not drawn to scale); y-axis depicts the average ensemble defect for the output of each software, on each target. (B) Average proportion of native (target) contacts, where the (non length-normalized) averages are taken over all sequences returned for a fixed target structure. Name of the target structure is given on the x-axis, arranged in increasing length (length is not drawn to scale); y-axis depicts the expected proportion of base pairs in the target structure that are present in the low energy ensemble of all structures, for each target. Benchmarking data, both raw data and length-normalized data as well as scatter plots for all measures can be found at the RNAiFold 2.0 web site in the ‘Download’ tab.
To our knowledge, the only other web servers or software for RNA design are: ERD (4), FRNAkenstein (3), Incarnation (19), Info-RNA (6), MODENA (2), NUPACK (1), RNA-SSD (web server is called RNAdesigner) (20), RNAfbinv (5), RNAinverse (14). Default parameters were used for all software, with the exception of RNAinverse, where we used flags −R −1. In addition to the features indicated in Table 1, RNAiFold (both versions 1.0 and 2.0) is to our knowledge the only software that allows the user to stipulate exact GC-content range for the sequences returned; although Incarnation is claimed to support GC-content range stipulation, it returns some sequences that violate the user-stipulated GC-content. RNAdesign allows the user to designate a ‘target’ GC-content; however, this only introduces a bias trying to adjust the GC-content of the output sequences.
RNAiFold 2.0 also allows the user to stipulate the energy range E(a, S0) for the sequences a returned, where S0 denotes the target structure. The ERD web server (http://mostafa.ut.ac.ir/corna/erd-cons/) claims to support energy range stipulation, but in a test only one of the 10 requested solutions for target structure ((((...)))). had energy in the requested range of −4 to −2 kcal/mol. RNAiFold 2.0 can as well return that sequence which folds into the target S0 and has minimum energy among all such sequences; however, this feature is not a true constraint and results from the fact that RNAiFold 2.0 generates all solutions. Apart from other features indicated in Table 1, NUPACK is the only software that solves the inverse folding problem for both DNA and RNA, and in the case of DNA, allows the user to stipulate magnesium and sodium ion concentrations.
To compare RNAiFold 2.0 with RNAiFold 1.0 and other existent inverse folding software, we used 63 target structures, ranging in size from 54 to 1398 and used in earlier benchmarking from (12) (data described later). For the comparison of versions 1.0 and 2.0 of RNAiFold, we ran each program 100 times per target structure, for each of the 63 target structures just described. Each run had a time upper bound of 10 min; however, execution was terminated as soon as the first solution was returned. A solution was returned on average within ∼10 s (10.78 s for RNAiFold 2.0, 12.64 s for RNAiFold 1.0). The web server RNAiFold 2.0 obtained more solutions than RNAiFold 1.0 for ∼10% of the data; i.e. 24 targets had more solutions, 21 targets had the same number and 18 targets had fewer solutions. RNAiFold 2.0 returned solutions in less time than RNAiFold 1.0 for ∼43% of the data; i.e. 38 targets were solved more quickly with version 2.0, 14 targets required the same time (or neither version returned a solution within 10 min), 11 targets were solved more quickly with version 1.0. Benchmarking data and target structures can be found on web server at the tab ‘Download’.
Table 2 illustrates differences in the quantity and quality of sequences returned for a given target structure. Measures that quantify the extent to which the ensemble of low energy structures of a given sequence resembles a target structure (ensemble defect, expected base pair distance) or how diverse structures are from each other (Morgan-Higgs and Vienna structural diversity) are defined in the next paragraph. For each of the 63 target structures, each software was run 10 min to generate a quantity of sequences using default settings. ERD returns an output 100% of the time, where 85% of the output sequences fold into the target structure. In contrast, RNAiFold 2.0 returns an output 65% of the time, but 100% of its output is guaranteed to fold into the target structure. Incarnation returns 41 535 sequences on average for each target, but less than 0.2% fold into the target structure, while RNAiFold 2.0 returns 55 476 sequences on average and 100% fold into the target structure. Info-RNA has over 72% GC-content, due to the initial choice of starting sequence, while NUPACK and RNAiFold 2.0 have around 57% GC-content (and moreover, RNAiFold 2.0 allows the user to set a desired GC-content range), while RNA-SSD has close to 36% GC-content.
Benchmarking data
All benchmarking data is available at http://bioinformatics.bc.edu/clotelab/RNAiFold2.0 (tab ‘Download’). It is comprised of dataset A from (2) and datasets B and C from (20). Dataset A consists of 29 target structures first described in (2), obtained by taking the Rfam structure of the longest sequence from the seed alignment for each of families RF00001-RF00030 in the Rfam 9.0 database (21), with the exception of family RF00023 (tmRNA). Dataset B consists of 24 target structures, whose GenBank accession codes are given and which were first described in (20); dataset C consists of 10 longer target structures described as follows—see (20) for references. (i) Minimal catalytic domain of the hairpin ribozyme satellite RNA from tobacco ringspot virus. (ii) U3 snoRNA 5′-domain from Chlamydomonas reinhardtii. (iii) 5S rRNA from Haloarcula marismortui. (iv) VS Ribozyme from Neurospora mitochondria. (v) R180 ribozyme. (vi) XS1 ribozyme, Bacillus subtilis P RNA-based ribozyme. (vii) RNase P RNA from Homo sapiens. (viii) S20 mRNA from Escherichia coli. (ix) RNAse P RNA from Halobacterium cutirubrum. (x) Domains 1,3,5 from the group II intron ai5γ from the mitochondria of Saccharomyces cerevisiae. (References for experimentally determined structures not given due to space constraints; see (6) for these references.)
RNA structural measures
A secondary structure S may be considered as the set of its base pairs, hence (i, j) is a base pair of S when (i, j) ∈ S and the collection of base pairs common to structures S, T is denoted S∩T. Many RNA structural measures can be defined from the base pairing probabilities, computed by McCaskill's algorithm (22) and implemented in RNAfold −p (14,23). Given the RNA sequence a = a1, …, an, let pi, j = ∑{S: (i, j) ∈ S}P(a, S) = ∑{S: (i, j) ∈ S}exp ( − E(a, S)/RT)/Z, where P(a, S) is the Boltzmann probability of structure S of RNA sequence a, E(a, S) is the Turner energy of secondary structure S (24,25), R ≈ 0.001987 kcal mol−1 K−1 is the universal gas constant, T = 310.15 is absolute temperature and the partition functionZ = ∑Sexp ( −E(a, S)/RT), where the sum is taken over all secondary structures S of a. Symmetrize the base pair probabilities, by defining pj, i = pi, j for 1 ≤ i, j ≤ n, i ≠ j and define pi, i = 1 − ∑i ≠ jpi, j to be the probability that position i is unpaired. Let s0 denote the MFE structure of input RNA sequence a.
Morgan-Higgs diversity (17) is defined for a = a1, …, an by
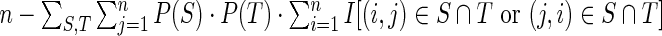 , where the sum is taken over all secondary structures S, T of a and I denotes the indicator function.
, where the sum is taken over all secondary structures S, T of a and I denotes the indicator function.Vienna diversity is defined for a = a1, …, an by ∑1 ≤ i < j ≤ npi, j(1 − pi, j) + (1 − pi, j)pi, j = ∑i < j2pi, j(1 − pi, j). In (26), this is called ensemble diversity.
Expected number of base pairs is defined by ∑1 ≤ i < j ≤ npi, j.
Expected base pair distance (9) is defined to the MFE structure s0 of input RNA sequence a is defined by
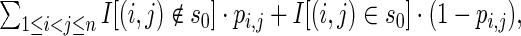 where I denotes the indicator function.
where I denotes the indicator function.Ensemble defect (18) is the expected number of nucleotides whose base pairing status differs from the target structure S0, defined by n − ∑i ≠ jpi, j · I[(i, j) ∈ S0] − ∑1 ≤ i ≤ npi, i · I[i unpaired in s0], where I denotes the indicator function.
Given sequence a = a1, …, an, the total positional entropy (H) (16) is defined by
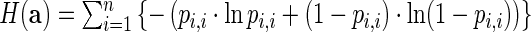 , where 0 · ln 0 is defined to be 0. Normalized positional entropy for a is H(a)/n; the average normalized positional entropy is the average normalized positional entropy, where the average is taken over all output sequences.
, where 0 · ln 0 is defined to be 0. Normalized positional entropy for a is H(a)/n; the average normalized positional entropy is the average normalized positional entropy, where the average is taken over all output sequences.Given the RNA sequence a = a1, …, an and target structure S0, the expected proportion of native (i.e. target) contacts
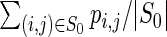 , where |S0| denotes the number
, where |S0| denotes the number 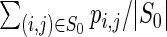 , where |S0| denotes the number of base pairs in S0. The average is then taken over all sequences output for a given target structure, and then the average is taken over all targets. This value is called ‘mean fraction of bases retained’ in (27), where it is approximated by sampling using RNAsubopt (14,28).
, where |S0| denotes the number of base pairs in S0. The average is then taken over all sequences output for a given target structure, and then the average is taken over all targets. This value is called ‘mean fraction of bases retained’ in (27), where it is approximated by sampling using RNAsubopt (14,28).
RESULTS
In this section, we illustrate how to use RNAiFold 2.0, in order to design synthetic RNAs that trigger two biologically significant recoding events: (i) SECIS elements cause the ribosome to incorporate a non-standard amino acid, selenocysteine, into a growing peptide chain; (ii) programmed −1 ribosomal frameshift signals cause the ribosomal reading frame in messenger RNA to shift at a specific site by −1 within the coding region.
Prokaryotes, archaea and eukaryotes employ the UGA stop codon to code for selenocysteine, rather than terminating protein translation, provided that a selenocysteine insertion (SECIS) element occurs downstream of the UGA stop codon. The SECIS element is a ∼42 nt sequence having conserved nucleotides at certain positions, which folds into a stem-loop secondary structure (29)—see target structure in Example 5. In prokaryotes, the SECIS element lies immediately after the UGA stop codon, while in eukaryotes and archaea it lies in the 3′ untranslated region (30). In the formate dehydrogenase F (fdhF) gene of Salmonella enterica (GenBank: CDS70432.2), the 42-nt sequence UGACACGGCC CAUCGGUUGC AGGUCUGCAC CAAUCGGUCG GU consists of the UGA stop codon immediately followed by the SECIS element. This sequence folds into the stem-loop structure shown in Example 6 and codes the 14 residue peptide UHGPSVAGLHQSVG (‘U’ denotes selenocysteine).
In contrast, the homologous 14 residue peptide of the fdhF protein of Raoultella ornithinolytica is given by CHGPSVAGLQQALG, where cysteine appears instead of selenocysteine. Unlike S. enterica, the 42 = 14 · 3 nt portion of the fdhF gene of R. ornithinolytica (Genbank AJF73661.1) begins with UGC, which codes for cysteine, rather than UGA, a stop codon which codes for selenocysteine in the presence of a SECIS element; moreover, the 42-nt sequence of R. ornithinolytica does not fold into a stem-loop SECIS structure.
The following input file defines the target structure to be the MFE structure of the 42-nt RNA from S. enterica, sets as sequence constraints the bulged U18 and GGUC hairpin identity (known to be important for SECIS functionality (31,32) and sets as amino acid constraints the 14-mer of R. ornithinolytica, with ‘C’ replaced by ‘U’.

In 0.24 s RNAiFold 2.0 determined the optimal solution UGACACGGGC CCUCGCUUGC AGGUCUGCAG CAAGCGCUCG GA, which begins by the UGA stop codon, translates the 14-mer UHGPSVAGLQQALG and folds into the requisite SECIS stem-loop. This example shows how RNAiFold 2.0 can be used to re-engineer selenoproteins from cysteine-bearing proteins.
In the retrovirus HIV-1, Pol is obtained from a fused Gag-Pol polyprotein via a programmed −1 ribosomal frameshift, which is caused by a heptameric slippery sequence (U UUU UUA), where the Gag reading frame is indicated, together with a downstream frameshift stimulating stem-loop structure (33). Using the target and constraints from Example 2, we ran RNAiFold 2.0 to find the complete set of 29 340 solutions in 539.49 s (≈9 h). The sequences returned by RNAiFold 2.0 constitute synthetic putative ribosomal frameshift signals, which could be tested for frameshift efficiency. Additionally, we can infer the relative importance of amino acid coding requirements for Gag and Pol versus secondary structure requirements within the frameshift signal, by comparing naturally occurring −1 ribosomal frameshift elements in Rfam family RF00480 with the solutions returned by RNAiFold 2.0.
CONCLUSION
RNAiFold 2.0 is a complete overhaul and reimplementation of the algorithms from (9) in C++ using the new OR-Tools engine https://code.google.com/p/or-tools/. Novel features of the new software and web server, beyond those of RNAiFold 1.0, include an automated pipeline for synthetic RNA design, use of Turner’99 or Turner’04 energy model, stipulation of a partial target structure, stipulation of prohibited (incompatible) base pairs and amino acid constraints. Given a target non-pseudoknotted hybridization complex of two structures, RNAiFold 2.0 can output pairs of sequences, whose MFE hybridization complex is equal to the target. All the previously described constraints are supported for hybridization—see the online manual section on Cofold for the syntax and an example. Availability of the source code will allow users to design synthetic RNAs, following the pipeline we used to design functional synthetic hammerhead ribozymes in (10).
Acknowledgments
We would like to thank anonymous reviewers for useful suggestions.
FUNDING
National Science Foundation [DBI-1262439]. Funding for open access charge: National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Zadeh J.N., Wolfe B.R., Pierce N.A. Nucleic acid sequence design via efficient ensemble defect optimization. J. Comput. Chem. 2011;32:439–452. doi: 10.1002/jcc.21633. [DOI] [PubMed] [Google Scholar]
- 2.Taneda A. MODENA: a multi-objective RNA inverse folding. Adv. Appl. Bioinform. Chem. 2011;4:1–12. doi: 10.2147/aabc.s14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyngso R.B., Anderson J.W., Sizikova E., Badugu A., Hyland T., Hein J. Frnakenstein: multiple target inverse RNA folding. BMC Bioinformatics. 2012;13:260. doi: 10.1186/1471-2105-13-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esmaili-Taheri A., Ganjtabesh M. ERD: a fast and reliable tool for RNA design including constraints. BMC Bioinformatics. 2015;16:20. doi: 10.1186/s12859-014-0444-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinbrand L., Avihoo A., Barash D. RNAfbinv: an interactive Java application for fragment-based design of RNA sequences. Bioinformatics. 2013;29:2938–2940. doi: 10.1093/bioinformatics/btt494. [DOI] [PubMed] [Google Scholar]
- 6.Busch A., Backofen R. INFO-RNA, A fast approach to inverse RNA folding. Bioinformatics. 2006;22:1823–1831. doi: 10.1093/bioinformatics/btl194. [DOI] [PubMed] [Google Scholar]
- 7.Hofacker I. Ph.D. Thesis. University of Vienna; 1994. The Rules of the Evolutionary Game for RNA: A A Statistical Characterization of the Sequence to Structure Mapping in RNA. [Google Scholar]
- 8.Gruber A., Lorenz R., Bernhart S., Neubock R., Hofacker I. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:70–74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Martin J.A., Clote P., Dotu I. RNAiFOLD: a constraint programming algorithm for RNA inverse folding and molecular design. J. Bioinform. Comput. Biol. 2013;11:1350001. doi: 10.1142/S0219720013500017. [DOI] [PubMed] [Google Scholar]
- 10.Dotu I., Garcia-Martin J.A., Slinger B.L., Mechery V., Meyer M.M., Clote P. Complete RNA inverse folding: computational design of functional hammerhead ribozymes. Nucleic Acids Res. 2015;42:11752–11762. doi: 10.1093/nar/gku740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dotu I., Lozano G., Clote P., Martinez-Salas E. Using RNA inverse folding to identify IRES-like structural subdomains. RNA Biol. 2013;10:1842–1852. doi: 10.4161/rna.26994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Martin J.A., Clote P., Dotu I. RNAiFold: a web server for RNA inverse folding and molecular design. Nucleic Acids Res. 2013;41:W465–W470. doi: 10.1093/nar/gkt280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner D.H., Mathews D.H. NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2010;38:D280–D282. doi: 10.1093/nar/gkp892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nawrocki E.P., Burge S.W., Bateman A., Daub J., Eberhardt R.Y., Eddy S.R., Floden E.W., Gardner P.P., Jones T.A., Tate J., et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–D137. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huynen M., Gutell R., Konings D. Assessing the reliability of RNA folding using statistical mechanics. J. Mol. Biol. 1997;267:1104–1112. doi: 10.1006/jmbi.1997.0889. [DOI] [PubMed] [Google Scholar]
- 17.Morgan S., Higgs P. Barrier heights between ground states in a model of RNA secondary structure. J. Phys. A Math. Gen. 1998;31:3153–3170. [Google Scholar]
- 18.Dirks R., Lin M., Winfree E., Pierce N. Paradigms for computational nucleic acid design. Nucleic Acids Res. 2004;32:1392–1403. doi: 10.1093/nar/gkh291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinharz V., Ponty Y., Waldispuhl J. A weighted sampling algorithm for the design of RNA sequences with targeted secondary structure and nucleotide distribution. Bioinformatics. 2013;29:i308–i315. doi: 10.1093/bioinformatics/btt217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andronescu M., Fejes A., Hutter F., Hoos H., Condon A. A new algorithm for RNA secondary structure design. J. Mol. Biol. 2004;336:607–624. doi: 10.1016/j.jmb.2003.12.041. [DOI] [PubMed] [Google Scholar]
- 21.Gardner P.P., Daub J., Tate J.G., Nawrocki E.P., Kolbe D.L., Lindgreen S., Wilkinson A.C., Finn R.D., Griffiths-Jones S., Eddy S.R., et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCaskill J. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 23.Hofacker I. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews D., Sabina J., Zuker M., Turner D. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 25.Xia T., SantaLucia J., Burkard M., Kierzek R., Schroeder S., Jiao X., Cox C., Turner D. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1999;37:14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 26.Gruber A.R., Bernhart S.H., Hofacker I.L., Washietl S. Strategies for measuring evolutionary conservation of RNA secondary structures. BMC Bioinformatics. 2008;9:122. doi: 10.1186/1471-2105-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pei S., Anthony J.S., Meyer M.M. Sampled ensemble neutrality as a feature to classify potential structured RNAs. BMC Genomics. 2015;16:35. doi: 10.1186/s12864-014-1203-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wuchty S., Fontana W., Hofacker I.L., Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–164. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 29.Commans S., Böck A. Selenocysteine inserting tRNAs: an overview. FEMS Microbiol. Rev. 1999;23:333–351. doi: 10.1111/j.1574-6976.1999.tb00403.x. [DOI] [PubMed] [Google Scholar]
- 30.Grundner-Culemann E., Martin G., Harney J.W., Berry M.J. Two distinct SECIS structures capable of directing selenocysteine incorporation in eukaryotes. RNA. 1999;5:625–635. doi: 10.1017/s1355838299981542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z., Reches M., Groisman I., Engelberg-Kulka H. The nature of the minimal ‘selenocysteine insertion sequence’ (SECIS) in Escherichia coli. Nucleic Acids Res. 1998;26:896–902. doi: 10.1093/nar/26.4.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandman K.E., Tardiff D.F., Neely L.A., Noren C.J. Revised Escherichia coli selenocysteine insertion requirements determined by in vivo screening of combinatorial libraries of SECIS variants. Nucleic Acids Res. 2003;31:2234–2241. doi: 10.1093/nar/gkg304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ofori L.O., Hilimire T.A., Bennett R.P., Brown N.W. Jr, Smith H.C., Miller B.L. High-affinity recognition of HIV-1 frameshift-stimulating RNA alters frameshifting in vitro and interferes with HIV-1 infectivity. J. Med. Chem. 2014;57:723–732. doi: 10.1021/jm401438g. [DOI] [PMC free article] [PubMed] [Google Scholar]