

Abstract
Identifying protein functions can be useful for numerous applications in biology. The prediction of gene ontology (GO) functional terms from sequence remains however a challenging task, as shown by the recent CAFA experiments. Here we present INGA, a web server developed to predict protein function from a combination of three orthogonal approaches. Sequence similarity and domain architecture searches are combined with protein-protein interaction network data to derive consensus predictions for GO terms using functional enrichment. The INGA server can be queried both programmatically through RESTful services and through a web interface designed for usability. The latter provides output supporting the GO term predictions with the annotating sequences. INGA is validated on the CAFA-1 data set and was recently shown to perform consistently well in the CAFA-2 blind test. The INGA web server is available from URL: http://protein.bio.unipd.it/inga.
INTRODUCTION
Although the biological role of a protein is encoded in its sequence, a simple function to map a protein sequence into its biological activity is unknown. Moreover, experimental techniques to determine protein function are costly and time consuming. Filling the gap between the number of available sequences and their functional characterization requires computational methods (1). The most widely used ontology to describe protein function is the Gene Ontology (GO) (2). GO defines three sub-ontologies (molecular function, biological process and cellular component) describing different aspects of function. GO terms are grouped in a hierarchical way as a directed acyclic graph, where deeper nodes correspond to more specialized functions.
Recently, the Critical Assessment of protein Function Annotation (CAFA) challenge has started to provide an objective overview of the state of the art in the field of automatic protein function prediction (3). The CAFA experiment was also responsible for defining some new criteria for evaluation, like the validation data set used for the blind test, the definition of function space through GO terms and scoring metrics for comparing different methods. Most available algorithms exploit homology inference to assign function (4–7). This is based on the logic that evolutionarily related proteins share a common ancestor from whom the function was inherited. However, it is very difficult to infer homology for highly divergent proteins, in particular when it is impossible to build a reliable phylogenetic tree (8). Sequence similarity alone is also not a sufficient condition to infer functional similarity, as the function of identical sequences may change depending on different in vivo environments such as organism, tissue or sub-cellular localization. Other methods exploit domain organization in the sequence to predict function (9–12). By definition, a protein domain corresponds to a functional unit and the combination of different units provides the cell with a way to develop new functions in a modular fashion (13). Annotation can then be transferred among proteins sharing the same domain architecture. The performance of these kinds of predictors relies on the ability to find domains in a given sequence as well as the quality of the functional annotation for the domains themselves. Other approaches involve the use of information available in protein–protein interaction (PPI) networks (14,15). The assumption is that whenever a protein physically interacts with other proteins, it is part of the same biological process and located in the same cellular compartment. Of course, this is not always true. For example, in the case of chaperones or ubiquitin interacting with a broad set of functionally unrelated protein partners. This assumption, however, holds in the majority of cases, as proteins belonging to the same pathway have been shown to be strongly interconnected (16). The main problem of methods based on PPI data is coverage, as it is impossible to make predictions whenever the interacting partners of a protein are not known. Thanks to the huge amount of data generated by new experimental techniques (17,18), information available in PPI networks has become more relevant. The coverage problem has become a minor issue and methods based on PPI data are promising. According to the results of the CAFA experiment (3), the best methods rely on consensus predictions and are in general able to exploit different sources of information (19,20).
Here we introduce INGA, Interaction Network GO Annotator, a tool that predicts protein function exploiting PPI networks, sequence similarity measures and domain assignments. INGA combines the three different component predictors to generate a consensus that outperforms them. It was recently evaluated at the second CAFA experiment (2014; URL: http://biofunctionprediction.org/) and ranked among the ten best methods both for molecular function and biological process prediction.
MATERIALS AND METHODS
INGA is a method that generates a consensus combining three different predictors. The different information sources are PPI networks, sequence similarity and domain assignments. In the web server, these components are identified as CONSENSUS, BLAST, PFAM and STRING to recall the information source. The consensus prediction provided by INGA has been evaluated in the CAFA-2 assessment resulting among the top ten methods. The accuracy of INGA arises from several implementation details. The most important factors are: choice of the network, strategy adopted to transfer annotation, approach to identify domains and the way homology is inferred. A description of the implemented predictors and the strategies adopted to maximize the accuracy of the combined consensus prediction follows. Of course, like all predictors, INGA is susceptible to systematic annotation errors in the source databases and the user should evaluate predictions carefully. To this end, INGA provides an interface to track all information sources and help the user estimate their reliability.
Protein interaction networks (STRING)
Proteins in a living cell have many physical interactors. Each group of interacting proteins is expected to participate in the same biological process and to operate in the same sub-cellular compartment. Proteins involved in the same pathway are indeed shown to be more interconnected (16). Thanks to an increasing amount of available interaction data, it is sensible to exploit this information to predict protein function, at least for Cellular Component and Biological Process terms. Given a protein target, INGA collects the set of directly interacting nodes from the STRING (21) database (v9.1). When the target sequence is not exactly present as STRING entry, INGA tries to find a similar entry with at least 90% sequence identity and 90% coverage. When the mapping is not one-to-one, INGA merges the set of interacting nodes of all mapped entries in a single group. UniProt (22) (release 2014_08) is then queried to retrieve all associated GO (2) terms from the set of interaction partners. For each GO term, its enrichment in the group of interacting proteins (sub network) compared to the rest of annotated nodes in the entire STRING network, considering all organisms together, is calculated. Enrichment is measured by calculating a P-value with Fisher's exact test and used to rank GO terms. The P-value represents the probability that a GO term is associated with a group of interactors by chance (null hypothesis) and is calculated with the formula below applied to a contingency table (see Table 1):
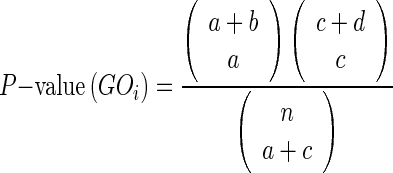 |
(1) |
Table 1. Contingency table for enrichment calculation. Subset indicates the set of directly interacting nodes in STRING and the set with the same architecture in Pfam. Rest represents the remaining nodes in STRING and proteins in UniProt with GO terms associated with Pfam domains.
| Data set | |||
|---|---|---|---|
| Subset | Rest | ||
| Categories | GOi | a | b |
| Not GOi | c | d | |
Domain assignments (PFAM)
Protein domains are independent folding units and represent the basic modules for protein function (13). It has been postulated that the evolution of proteins can explain the complexity of the cell through rearrangement of fragment units and different combinations of a relatively small number of domains (23). The basic idea in INGA is to exploit the domain architecture and transfer annotation from proteins sharing the same domain pattern. Given a protein sequence, INGA identifies putative Pfam (24) domains using HMMER (25). All proteins with the same set of domains are retrieved from UniProt and the associated GO annotation is transferred to the target. GO terms are ranked with the same approach adopted for the protein interaction network (see previous paragraph). The enrichment of each GO term in the group of proteins with the same domain architecture is compared to the distribution of GO terms in the rest of the database (see Table 1). The P-value associated to a GO term represents the null hypothesis that the GO term is associated with the group by chance and is calculated with the same formula used for STRING enrichment.
Sequence similarity (BLAST)
According to the CAFA evaluation, the best predictors used sequence similarity to infer homology and predict function (6,19,20). All use either Blast (26) or PsiBlast (27) to find similar proteins. The BLAST predictor implemented in INGA, exploits the ‘transfer by homology’ principle, and transfers GO terms from sequences that are considered evolutionarily related on the basis of their sequence similarity. In particular, INGA considers it safe to transfer function only when BLAST matches proteins that share more than 40% sequence identity with a coverage (alignment overlap) greater than 80%. The search is performed against the entire UniProt sequence database (release 2014_08). Sequences with an e-value higher than 10−3 were excluded. The coverage constraint guarantees to exclude sequence pairs that align well locally for a fragment that may correspond to a single domain while the rest of the sequence remains unaligned. The list of retrieved hits is then filtered a second time, removing hits lacking either any GO annotation or experimental annotation according to the user choice. Valid hits were sorted by the Blast bit-score and GO terms were sorted accordingly. It has been shown that the bit-score provides a better sorting compared to sequence identity when evaluating the function of retrieved hits (3,19).
Consensus and training
Consensus methods result particularly effective when combining predictors that exploit orthogonal information sources which provide coherent results. Our case fits this situation well, since interaction data can be considered independent from homology inference based on sequence similarity and domain assignments. The consensus is calculated to maximize the F-score (or F-measure) as used in CAFA (3). The F-score represents the quality of a given prediction and is the harmonic mean between precision and recall. It can be used as a probability, with higher probabilities corresponding to better predictions. If Pm represents the probability (confidence score) of the term GOi given by the method m, the consensus score is calculated as a joint probability with the following formula:
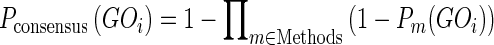 |
(2) |
Pconsensus is higher when a term is predicted by multiple predictors. For each method and each GO term, we generate Pm with the following formula:
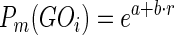 |
(3) |
Where e is Euler's number, a and b are two parameters and r represents the ranking, i.e. the position of GOi in the output list for method m. Rank 1, for BLAST, corresponds to all GO terms (plus ancestors) associated with the first hit (i.e. best bit-score), while in STRING and PFAM rank 1 identifies the terms with the lowest P-value. We decided to generate probabilities with the ranking instead of raw scores (P-value and bit-score) as they correlate much better with the F-score (data not shown). Moreover, equation 3 is very simple and requires only the a and b parameters to be estimated. Training has been performed by simply fitting the parameters of the ranking position to maximize the F-score in the training set. A total of 36 models were generated reflecting different predictors (BLAST, PFAM, four versions of STRING based on different edge confidence), different annotation sources (experimental and all annotations) and different sub-ontologies (Molecular Function, Biological Process and Cellular Component). Parameters for all models were optimized on a training set of 10 000 experimentally annotated SwissProt proteins (release 2013_07), as defined in CAFA, with GO terms associated with trusted evidence codes: inferred from Experiment (EXP), inferred from Direct Assay (IDA), inferred from Physical Interaction (IMP), inferred from Genetic Interaction (IGI), inferred from Expression Pattern (IEP), Traceable Author Statement (TAS) and Inferred by Curator (IC).
Evaluation
In the past, the evaluation of a function prediction method was usually carried out by comparing the annotation of model organisms like Yeast on specific ad hoc ontologies which were much smaller than the current GO. Moreover, it is very difficult to fairly evaluate the state of the art. Most methods predict annotation by transferring information from public annotation databases and by this are very sensitive to the ability of exploiting updated data. The CAFA experiment solves these problems by introducing a fair blind test. All participants are asked to predict GO terms for a set of sequences (validation set). After usually six months, CAFA closes the submission phase and starts to collect experimental annotation for another six months. The evaluation is then performed on the sequence subset that gained GO terms in the annotation phase. The CAFA evaluation is mainly based on precision-recall curves and other metrics described here (3). INGA, identified as Tosatto-UniPD, has been ranked among the ten best predictors both for Molecular Function and Biological Process sub ontologies in the second CAFA edition (2014; URL: http://biofunctionprediction.org/). The official assessment paper has not yet been published at the time of writing. Therefore, here we provide an evaluation based on the test set of the first CAFA edition. We simulated the same blind test by generating INGA predictions using the data and ontology available before the submission deadline (18 January 2011). The parameters of equation 3, a and b, were trained by extracting a subset of 10 000 experimentally annotated proteins randomly selected from SwissProt (release 2010_12). Table 2 shows the INGA performance in terms of F-score compared with the BLAST and NAIVE methods implemented as described in CAFA. INGA predictions are always better both for precision and F-score. Comparing our table with the CAFA results, INGA is ranked among the top five methods. It should be noted that, compared to the official CAFA-1 evaluation, the BLAST and NAIVE F-scores are slightly different. This is probably due to differences in the implementation as some details are missing in the published paper (e.g. release of the annotation database, ontology version and some BLAST parameters). To generate predictions we used UniProt 2011_01, the gene ontology of the 1st January 2011 and BLAST with the following parameters: -num_alignments 250 and -evalue 0.01. All INGA predictions for the CAFA-1 set are available for download from URL http://protein.bio.unipd.it/inga/INGA-evaluation.tar.gz.
Table 2. CAFA-1 validation set performance. Precision, recall and F-score values are shown at the confidence threshold maximizing the F-score for each method. In each column the highest value is highlighted in bold. The confidence values are sequence identity for BLAST, term frequency in the experimental annotation database for NAIVE and CONSENSUS score for INGA. Coverage represents the fraction of target proteins for which a method predicts at least one GO term. For NAIVE the coverage is always 1 by definition and not reported.
| Confidence threshold | Coverage | Precision | Recall | F-score | ||
|---|---|---|---|---|---|---|
| Molecular Function | BLAST | 0.25 | 0.64 | 0.37 | 0.47 | 0.42 |
| NAIVE | 0.19 | 0.29 | 0.23 | 0.26 | ||
| INGA | 0.80 | 0.91 | 0.53 | 0.63 | 0.58 | |
| Biological Process | BLAST | 0.25 | 0.76 | 0.18 | 0.42 | 0.25 |
| NAIVE | 0.20 | 0.28 | 0.21 | 0.24 | ||
| INGA | 0.78 | 0.80 | 0.37 | 0.33 | 0.35 | |
| Cellular Component | BLAST | 0.27 | 0.81 | 0.35 | 0.61 | 0.45 |
| NAIVE | 0.27 | 0.41 | 0.48 | 0.44 | ||
| INGA | 0.65 | 0.96 | 0.42 | 0.58 | 0.49 |
Implementation
The INGA web server is implemented using the REST (Representational State Transfer) architecture. The INGA services can be accessed both from a web interface and a Python API implemented ad hoc. The submitted job can be retrieved at a later time by providing the session identifier or the URL to the result page. INGA guarantees to maintain job sessions for at least two weeks. Predictions are stored permanently in a database where entries are indexed by their sequence in order to speed up the service when requesting a cached protein. Moreover INGA takes advantage of an implementation that allows to run the three different predictors in parallel, with results appearing independently as soon as a predictor finishes.
SERVER DESCRIPTION
Input
The INGA user interface is straightforward to use. The main page features a search box, which accepts any valid UniProt accession code (i.e. Q9XYF4) or, alternatively, Fasta sequence. Up to 10 input sequences or identifiers can be provided for each job. An optional title can be set to help the user distinguish between different prediction runs. Only two parameters need to be set for function prediction, the annotation database and the STRING edge confidence. The annotation database parameter refers to the way GO terms are filtered. Selecting the ‘Experimental’ button, only GO terms associated with trusted evidence codes will be selected, otherwise all GO terms from UniProt will be included without any filter (i.e. including electronically inferred). For the STRING edge confidence parameter, four confidence levels for interaction partner prediction can be selected. A high confidence score reduces the number of interacting partners, implying fewer sources for GO terms transfer and presumably fewer false positives. The default settings are ‘experimental’ annotations and ‘medium’ STRING edge confidence. This works well in most cases. For function prediction of proteins with very poor annotation, we recommend to use all database sources and low level of confidence on STRING interactors.
Output
INGA provides output for all the three implemented predictors and the consensus on the same page (see Figure 1). Each column lists predictions from one method and the GO terms are grouped by sub-ontology (molecular function; biological process; cellular component) to facilitate comparison between different predictors. By default, the tool displays only the first three predicted terms for each sub-ontology, but it is possible to expand the list and visualize all predicted annotations by clicking on the ‘Expand’ button. GO terms in the expanded list are grouped by score (rank) and sorted by term specificity. The block of terms with the same score is highlighted by a colored line on the left. The background color of each box reflects the informativeness of each term. More specific terms (i.e. farther away from the GO root) have darker background color and are always on the top of a score block. For each block, except consensus, the annotation source (UniProt accession codes) is also reported. For Pfam it is also possible to see the set of domains identified in the input sequence and matched against UniProt proteins. The list of GO terms associated with each sub-ontology and the prediction sources can be saved in an output file for download by clicking on the ‘download’ button on the top right corner.
Figure 1.
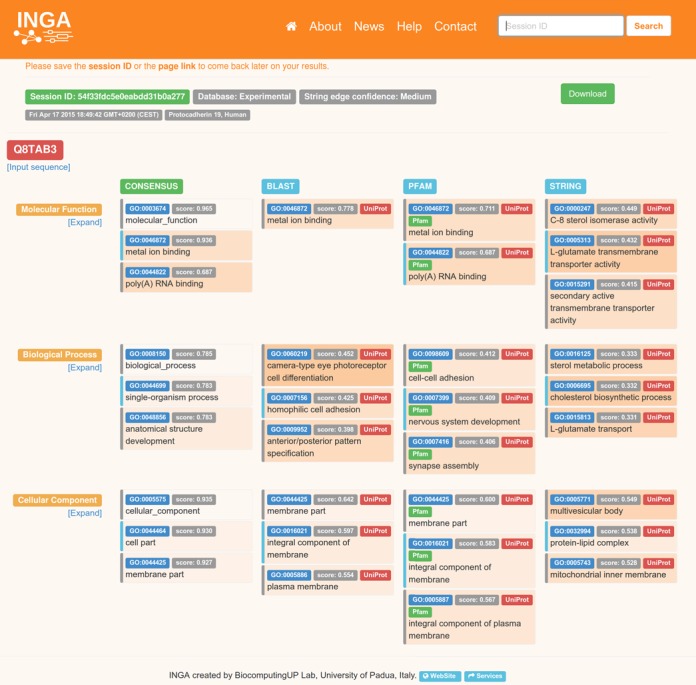
INGA results page for the human PCDH19 protein. The output contains a short header, with the session identifier, optional job title, download button as well as the UniProt accession code or title of the Fasta file used as input. The following three sections cover the GO sub-ontologies for molecular function, biological process and cellular component. Inside each section, the first column represents the consensus and is followed by BLAST, PFAM and STRING results. Each component result lists the GO term identifier, score and GO term description inside a box of variable background color. Darker background colors correspond to more informative (i.e. deeper) GO levels. Scores are in the range 0.0 (low) to 1.0 (high), with 1.0 reserved for curated GO terms for the query protein retrieved from UniProt. Where possible, UniProt and Pfam buttons open pop-up windows listing the UniProt accession numbers supporting the predicted GO term. An alternating gray or light blue stripe on the left side of the results box indicates whether consecutive entries belong to the same prediction or not. This is important, as multiple GO tracing the way from the prediction back to the ontology root can be shown. By default, only the top three results are shown for each GO sub-ontology and method. Clicking on ‘Expand’ below the GO sub-ontology name will expand the results section to cover all informative predictions for each method. Clicking on ‘Expand all’ will increase the number of visualized predictions further by listing all parent nodes of each prediction.
Usage example
Protocadherin-19 (PCDH19) is a member of the protocadherin (pcdhs) subfamily within the large cadherin superfamily, which were first discovered as calcium dependent cell-cell adhesion molecules involved in early vertebrate embryo development (28). Mutations in the PCDH19 gene are responsible for X linked, female-limited epilepsy and mental retardation (29,30). PCDH19 is highly expressed in the central nervous system but its cellular role is poorly understood. In public protein sequence databases, PCDH19 is simply described as a calcium binding molecule which mediates homophilic cell-cell adhesion. We used INGA to retrieve further information on PCDH19 protein function using the UniProt accession code (Q8TAB3) as input and only experimental sources for GO terms selection (Figure 1). Medium confidence interaction partner prediction has been chosen as starting condition for the analysis. The results page reports the four lists of predictions for each method. The first three GO terms with highest score obtained from the consensus method correctly predicted PCDH19 as a membrane protein binding calcium ions which is involved in cell-cell adhesion. The prediction is inferred from both homologous sequences and proteins sharing the same cadherin domain. Expanding the lists we can find other interesting GO terms associated with PCDH19. In particular, from the consensus prediction this protein is specifically predicted to be localized in neuron parts. The source of this prediction can be found searching for GO terms related to ‘neuron’ in the PFAM and STRING lists. Interestingly, the GO term refers to experimental data on proteins belonging to different pcdhs subfamilies. Recently, it has been show that pcdhs have an active role in neural circuit formation, recruiting regulators of cytoskeletal dynamics to the cell surface at interaxonal contact sites to induce persistent cell motility (31). Cellular component movement is also predicted as biological process in which PCDH19 is involved. This is also reported in literature where Biswas and colleagues demonstrated that PCDH19 interacts with NCAD to regulate cell adhesion and movement during anterior neurulation in zebra fish (32).
CONCLUSIONS
We have presented INGA, a novel method to predict protein function from sequence. It was optimized to combine three orthogonal sources of information, PPI networks, domain architecture and sequence similarity, into a consensus prediction for each of the three GO sub-ontologies. INGA performed consistently well at the most recent CAFA experiment. The web server was carefully designed to provide users with the necessary information to evaluate the biological meaning of the predicted functional terms. We anticipate that it will be useful both for large-scale annotation efforts, through its RESTful web services, and experimental biologists interested in designing experiments to test the function of a specific protein.
Acknowledgments
The authors are grateful to Emilio Potenza for initial help with the web server and to members of the BioComputing UP group for insightful discussions.
FUNDING
FIRB Futuro in Ricerca [RBFR08ZSXY]; University of Padua [CPDR123473]; AIRC [MFAG12740 to S.T.]; Italian Ministry of Health [GR-2011-02346845 to S.T. and GR-2011-02347754 to E.L.]; FIRC Fondazione Italiana per la Ricerca sul Cancro [Project No. 16621 to D.P.]. Funding for open access charge: FIRB Futuro in Ricerca.
Conflict of interest statement. None declared.
REFERENCES
- 1.Friedberg I. Automated protein function prediction–the genomic challenge. Brief. Bioinform. 2006;7:225–242. doi: 10.1093/bib/bbl004. [DOI] [PubMed] [Google Scholar]
- 2.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radivojac P., Clark W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A., et al. A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013;10:221–227. doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loewenstein Y., Raimondo D., Redfern O.C., Watson J., Frishman D., Linial M., Orengo C., Thornton J., Tramontano A. Protein function annotation by homology-based inference. Genome Biol. 2009;10:207. doi: 10.1186/gb-2009-10-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piovesan D., Luigi Martelli P., Fariselli P., Zauli A., Rossi I., Casadio R. BAR-PLUS: the Bologna Annotation Resource Plus for functional and structural annotation of protein sequences. Nucleic Acids Res. 2011;39:W197–W202. doi: 10.1093/nar/gkr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piovesan D., Martelli P.L., Fariselli P., Profiti G., Zauli A., Rossi I., Casadio R. How to inherit statistically validated annotation within BAR+ protein clusters. BMC Bioinformatics. 2013;14(Suppl. 3):S4. doi: 10.1186/1471-2105-14-S3-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chitale M., Hawkins T., Park C., Kihara D. ESG: extended similarity group method for automated protein function prediction. Bioinformatics. 2009;25:1739–1745. doi: 10.1093/bioinformatics/btp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelhardt B.E., Jordan M.I., Srouji J.R., Brenner S.E. Genome-scale phylogenetic function annotation of large and diverse protein families. Genome Res. 2011;21:1969–1980. doi: 10.1101/gr.104687.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter S., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bork P., Das U., Daugherty L., Duquenne L., et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dessailly B.H., Redfern O.C., Cuff A., Orengo C.A. Exploiting structural classifications for function prediction: towards a domain grammar for protein function. Curr. Opin. Struct. Biol. 2009;19:349–356. doi: 10.1016/j.sbi.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Lima Morais D.A., Fang H., Rackham O.J.L., Wilson D., Pethica R., Chothia C., Gough J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011;39:D427–D434. doi: 10.1093/nar/gkq1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rentzsch R., Orengo C.A. Protein function prediction using domain families. BMC Bioinformatics. 2013;14(Suppl. 3):S5. doi: 10.1186/1471-2105-14-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 14.Brun C., Chevenet F., Martin D., Wojcik J., Guénoche A., Jacq B. Functional classification of proteins for the prediction of cellular function from a protein-protein interaction network. Genome Biol. 2003;5:R6. doi: 10.1186/gb-2003-5-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chua H.N., Sung W.-K., Wong L. Using indirect protein interactions for the prediction of Gene Ontology functions. BMC Bioinformatics. 2007;8(Suppl. 4):S8. doi: 10.1186/1471-2105-8-S4-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barabási A.-L., Gulbahce N., Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho Y., Gruhler A., Heilbut A., Bader G.D., Moore L., Adams S.-L., Millar A., Taylor P., Bennett K., Boutilier K., et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 18.Johnson D.S., Mortazavi A., Myers R.M., Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 19.Cozzetto D., Buchan D.W.A., Bryson K., Jones D.T. Protein function prediction by massive integration of evolutionary analyses and multiple data sources. BMC Bioinformatics. 2013;14(Suppl. 3):S1. doi: 10.1186/1471-2105-14-S3-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fontana P., Cestaro A., Velasco R., Formentin E., Toppo S. Rapid annotation of anonymous sequences from genome projects using semantic similarities and a weighting scheme in gene ontology. PLoS One. 2009;4:e4619. doi: 10.1371/journal.pone.0004619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore A.D., Björklund A.K., Ekman D., Bornberg-Bauer E., Elofsson A. Arrangements in the modular evolution of proteins. Trends Biochem. Sci. 2008;33:444–451. doi: 10.1016/j.tibs.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 24.Punta M., Coggill P.C., Eberhardt R.Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul S. Basic Local Alignment Search Tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 27.Altschul S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyatani S., Shimamura K., Hatta M., Nagafuchi A., Nose A., Matsunaga M., Hatta K., Takeichi M. Neural cadherin: role in selective cell-cell adhesion. Science. 1989;245:631–635. doi: 10.1126/science.2762814. [DOI] [PubMed] [Google Scholar]
- 29.Dibbens L.M., Tarpey P.S., Hynes K., Bayly M.A., Scheffer I.E., Smith R., Bomar J., Sutton E., Vandeleur L., Shoubridge C., et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 2008;40:776–781. doi: 10.1038/ng.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leonardi E., Sartori S., Vecchi M., Bettella E., Polli R., Palma L.D., Boniver C., Murgia A. Identification of Four Novel PCDH19 Mutations and Prediction of Their Functional Impact. Ann. Hum. Genet. 2014;78:389–398. doi: 10.1111/ahg.12082. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi S., Inoue Y., Kiyonari H., Abe T., Misaki K., Moriguchi H., Tanaka Y., Takeichi M. Protocadherin-17 mediates collective axon extension by recruiting actin regulator complexes to interaxonal contacts. Dev. Cell. 2014;30:673–687. doi: 10.1016/j.devcel.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Biswas S., Emond M.R., Jontes J.D. Protocadherin-19 and N-cadherin interact to control cell movements during anterior neurulation. J. Cell Biol. 2010;191:1029–1041. doi: 10.1083/jcb.201007008. [DOI] [PMC free article] [PubMed] [Google Scholar]