

Abstract
i-cisTarget is a web tool to predict regulators of a set of genomic regions, such as ChIP-seq peaks or co-regulated/similar enhancers. i-cisTarget can also be used to identify upstream regulators and their target enhancers starting from a set of co-expressed genes. Whereas the original version of i-cisTarget was focused on Drosophila data, the 2015 update also provides support for human and mouse data. i-cisTarget detects transcription factor motifs (position weight matrices) and experimental data tracks (e.g. from ENCODE, Roadmap Epigenomics) that are enriched in the input set of regions. As experimental data tracks we include transcription factor ChIP-seq data, histone modification ChIP-seq data and open chromatin data. The underlying processing method is based on a ranking-and-recovery procedure, allowing accurate determination of enrichment across heterogeneous datasets, while also discriminating direct from indirect target regions through a ‘leading edge’ analysis. We illustrate i-cisTarget on various Ewing sarcoma datasets to identify EWS-FLI1 targets starting from ChIP-seq, differential ATAC-seq, differential H3K27ac and differential gene expression data. Use of i-cisTarget is free and open to all, and there is no login requirement. Address: http://gbiomed.kuleuven.be/apps/lcb/i-cisTarget.
INTRODUCTION
The field of regulatory genomics is generating vast amounts of sequencing data related to transcription factor binding, chromatin activity and gene expression. Whereas many tools are available for the functional analysis of gene signatures, such as Gene Ontology enrichment analysis (1–3) and for the identification of enriched transcription factor motifs in co-expressed gene sets (4–8), fewer web tools exist to analyse sets of genomic regions. Different types of post-processing and functional analysis of a set of genomic regions can be used to gain insights into regulatory and functional relationships. Firstly, motif discovery identifies transcription factor binding sites and predicts new regulators and co-factors. Tools exist for de novo motif discovery (e.g. PeakMotifs (9), MEME (10)) and for the enrichment analysis using libraries of position weight matrices (e.g. oPOSSUM-3 (4), the SeqPos tool in Cistrome (11) and Homer (7), although the latter is only available command-line). A second question one can ask for an experimentally derived set of genomic regions is whether it correlates with existing ChIP-seq or chromatin activity data such as histone modifications or open chromatin (DNaseI hypersensitivity, FAIRE-seq, ATAC-seq). An example tool that performs such correlations is the ENCODE ChIP-Seq Significance Tool (12). A third kind of analysis that is often performed on genomic regions is to associate each region to one or more candidate target genes and analyse the function (e.g. by GO (13)) of the resulting target gene set. Such a procedure is implemented by the web tool GREAT (14).
i-cisTarget aims at combining motif and track enrichment in a single analysis through a unified statistical framework and goes beyond existing tools concerning the amount of candidate position weight matrices and the number of experimental data tracks tested. In this article we present a major update of i-cisTarget, now including support for human and mouse datasets; increasing our motif collection to nearly 10,000 PWMs; and adding human and mouse specific databases with more than 4000 regulatory data tracks. One of the challenges is to make these analyses computationally tractable, so that they can be run in a web tool. To this end, we generated collections of candidate regulatory regions (CRRs) for the human and mouse genome. These regions are scored and ranked offline, so that the online recovery analysis becomes highly efficient. The output of i-cisTarget are predictions of key transcription factors alongside a prioritized list of direct transcriptional targets and the actual cis-regulatory modules (CRM) and transcription factor binding sites.
MATERIALS AND METHODS
Regulatory regions and data sources
Defining candidate regulatory regions for the human, mouse and fly genome
We defined sets of CRRs for the human, mouse and fly genomes. To delineate human CRRs the following publicly available regulatory data were used (see Table 1A): DNAseI Hypersensitive (DHS) uniform clustered peaks across 125 cell lines from ENCODE (15), General Binding Preference models (16), regulatory elements from ORegAnno (17), VistaEnhancers (18), predicted cis-regulatory modules (19), CpG islands and proximal promoters (both downloaded from UCSC table browser (20)), conserved non-coding sequences (CNS) and ultraconserved elements (UCR). For mouse CRRs the same features (mouse genome) were used except General Binding Preference models, using ultra-conserved non-coding elements (21). DHS peaks in mouse cell lines were used (22) (Table 1B). Where needed the UCSC liftover tool (23) was used to convert genome coordinates to hg19 and mm9.
Table 1. Publicly available regulatory datasets used to create i-cisTarget human CRRs (A) and publicly available regulatory datasets used to create i-cisTarget mouse CRRs (B).
| A. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| GBP | CpG | Proximal promoters | CNS | UCR | Oreganno | Vista enhancers | CRMs | DHS | |
| Number of regions | 61550 | 27718 | 34722 | 232101 | 15931 | 23112 | 1339 | 123500 | 1281988 |
| % of the genome | 1.77 | 0.73 | 0.67 | 2.25 | 0.13 | 0.39 | 0.07 | 2.05 | 13.36 |
| B. | |||||||||
| CpG | Proximal promoters | CNS | UCR | Oreganno | Vista enhancers | CRMs | UCNE | DHS | |
| Number of regions | 16026 | 22984 | 231478 | 15927 | 16976 | 339 | 91176 | 4335 | 14971709 |
| % of the genome | 0.40 | 0.41 | 2.49 | 0.14 | 0.38 | 0.02 | 1.66 | 0.05 | 29.16 |
Next, all these features were merged and regions having an overlap of at least 20% with insulator elements or at least 80% of coding exons were removed. Next, regions with an overlap <20% with insulators or 80% with exons are split and the regions containing the insulator or coding exons were removed. Remaining regions are then filtered based on size and regions shorter than 30 bp are removed. Finally, any resulting regions shorter than 1000 bp were extended if possible to 1000 bp in a direction that prevents overlap with an insulator or exon. The complete procedure of creating CRRs yielded 1,223,024 and 938,376 regions for human and mouse, respectively (representing ∼35% of the human genome and ∼42% of mouse genome). The sizes of the regions are relatively short, with more than 96% of human and 93% of mouse regions being smaller or equal than 1000 bp. The procedure of creating the fly CRRs (136,353 regions) is described in (24). The sets of CRRs per species are provided as bed files on the i-cisTarget website.
Mapping user input to candidate regions
The input can be either a set of co-regulated genomic loci, such as ChIP-seq or DHS/FAIRE/ATAC-seq peaks; or a set of co-expressed genes. In both cases the input is linked to the CRRs (as shown in the Figure 1c and d). Peaks/loci are converted to the overlapping CRRs according to the ‘fraction of overlap’ parameter (specified by the user) representing the overlap between the input peaks and i-cisTarget CRRs. For example, if the fraction of overlap is set to 0.4 (default) then only i-cisTarget regions that overlap at least 40% with the input peaks will be used in the analysis. On the Report page we provide a text file with the overlap details of the input regions with i-cisTarget CRRs. If the input is a set of genes, then the input genes are linked to CRRs by collecting all CRRs located in the neighbourhood of a gene. This neighbourhood extends upstream and downstream of the transcription start site (TSS) and is a parameter that can be selected by the user. For human and mouse the default space is 20 kb around TSS and for fly it is set to 5 kb upstream of TSS and all introns.
Figure 1.
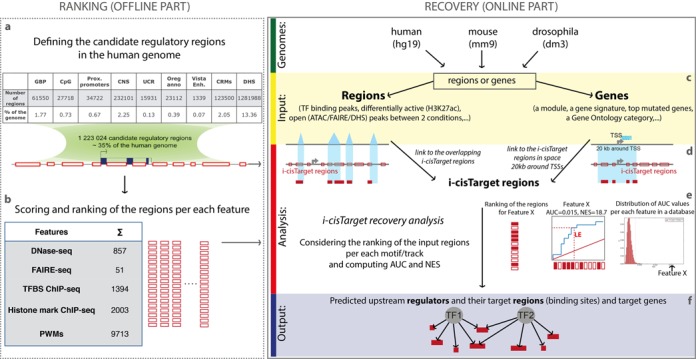
i-cisTarget workflow. The i-cisTarget web-tool consists of two parts, namely the ranking (offline part) and recovery (online part). (a) A set of 1,223,024 candidate regulatory regions (CRRs) is defined based on publicly available regulatory data, representing 35% of the human genome. (b) The collection of CRRs is scored and ranked according to different features, including motifs, TF and histone ChIP-seq, DNase-seq and FAIRE-seq, resulting in large ranking databases. (c) The online part starts with user input, which can be a set of genomic regions or a set of genes for human, mouse or fly. (d) The input set is mapped to the candidate i-cisTarget regions. In the case of regions/peaks, the overlapping CRRs (minimum overlap percentage is a parameter) with the peaks are considered in the analysis. When a gene set is used as input, then CRRs overlapping the entire space of X kb around TSSs are taken into the analysis (the default space for human and mouse genome is 20 kb around TSS; the default for fly is 5 kb upstream of TSS and all introns). (e) The recovery analysis identifies the feature for which the input CRRs are most enriched in the top of the CRR ranking of that feature. This enrichment is calculated by the Area Under the recover Curve (AUC) and all features with normalized AUC (i.e. Normalized Enrichment Score, or NES) above 3.0 are returned. (f) For each enriched feature and upstream regulator the direct target regions are provided, with a link to a BED file for download and a track in the UCSC Genome Browser.
Region-scoring and ranking
Scoring of candidate regulatory regions with position weight matrices
The motif collection was compiled from several resources which are described in (8) and yielded a large collection of 9713 position weight matrices (PWMs or motifs). The predefined CRRs were scored using Cluster-Buster (25) for each PWM across 10 mammalian genomes, namely: hg19 (Homo Sapiens), bosTau4 (Bos Taurus), canFam2 (Canis familiaris), mm9 (Mus musculus), monDom5 (Monodelphis domestica), panTro2 (Pan troglodytes), ponAbe2 (Pongo pygmaeus abelii), rheMac2 (Macaca mulatta), rn4 (Rattus norvegicus) and susScr2 (Sus scrofa). The orthologous regions between mammalian genomes were obtained using liftOver tool (23). This process is similar to the one used for motif scoring of fly CRRs as described in (24) and for human/mouse genes in the iRegulon Cytoscape plugin (8). Regions were ranked for each genome separately and subsequently the rankings were aggregated into one final ranking per PWM using OrderStatistics (26,27).
Scoring of candidate regulatory regions with experimental data tracks
Collections of human and mouse CRRs were also scored by the regulatory features: DHS, FAIRE, TF and histone ChIP-seq from ENCODE (15), Roadmap Epigenomics Project (28), data generated by Taipale laboratory (29), Aerts laboratory (H3K27ac, H3K27me3, FAIRE and MITF ChIP-seq on short-term melanoma cultures generated in-house (30) as well as p53 ChIP-seq and FAIRE on MCF-7 (8)) and Mouse ENCODE (22). Used feature types and resources are mentioned in Tables 2 A and B. Concerning the tracks from ENCODE, all the available replicates were used. For the scoring the maximum score of broad or narrow peaks (signalValue column in bed file format) within a region was used. Finally, each region has one score per track. Taipale datasets were converted from hg18 to hg19 using liftOver tool (23) and fold enrichment value was used for the ranking. Bigwig files were used for the ranking of the CRRs for tracks from Roadmap Epigenomics Project. Finally, three separate databases of rankings were created: DHS and FAIRE, histone modifications and TF ChIP-seq.
Table 2. Human regulatory tracks included in the databases (A) and mouse regulatory tracks included in the databases (B).
| A. | |||||
|---|---|---|---|---|---|
| ENCODE | Epigenome roadmap | Taipale | Aerts | ∑ | |
| DHS | 467 | 390 | 0 | 0 | 857 |
| FAIRE | 37 | 0 | 0 | 14 | 51 |
| Histone | 402 | 1572 | 3 | 26 | 2003 |
| TF ChIP-seq | 1274 | 0 | 117 | 3 | 1394 |
| ∑ | 2180 | 1962 | 120 | 43 | 4305 |
| B. | |||||
| ENCODE | |||||
| DHS | 150 | ||||
| FAIRE | 0 | ||||
| Histone | 209 | ||||
| TF ChIP-seq | 206 | ||||
| ∑ | 565 | ||||
Recovery analysis
The i-cisTarget enrichment analysis is based on calculating the cumulative recovery of the input regions along each ranking in the selected i-cisTarget databases, similarly to gene set enrichment analysis (GSEA)-like approaches (31). Briefly, the rank positions of the user input (mapped to i-cisTarget CRRs) is considered for each motif/track ranking and the Area Under the recovery Curve (AUC) of these foreground regions is calculated. We are mainly interested in regions that are highly ranked and therefore we calculate the AUC for only a fraction of the top ranked regions. This fraction is defined by AUC threshold parameter, which is set by default at 0.5% (6115 and 4692 regions) for human and mouse, at 1% for fly genome (1364 regions). The raw AUCs are then normalized to a Normalized Enrichment Score: NES = (AUC-μ)/σ, where μ represents the mean of all AUC scores across all features in the corresponding database and σ represents standard deviation of all these AUC scores. Finally, the optimal threshold (or ‘leading edge’) in the recovery curve represents the optimal set of target regions and corresponds to the rank position (x-axis) at which the difference of recovered regions (y-axis) with the average number of recovered regions plus two standard deviations is largest; this is illustrated in Figure 1e. In this way the input set is divided into true positive sets (sets of cis-regulatory elements where upstream regulators bind; the input regions within the LE subset) and false positives (the input regions outside the LE subset). Note that since the analysis based on the entire PWM database can take several minutes (also depending on the input dataset), we implemented an option to run a quick analysis, based on the JASPAR motif collection (1312 PWMs) (32) and a more stringent NES threshold (NES > 4.0). These quick jobs are prioritized in the job queue so that preliminary results can usually be obtained within ∼20 s, whereas a full job may take around 5 min.
Motif2TF mapping and motif clustering
Motif2TF mapping is used to associate the enriched motifs with candidate transcription factors. This mapping is based on a motif-TF network including direct evidence, orthology and motif–motif similarity as described in (8). Moreover, similar enriched motifs are clustered together by STAMP (33) which measures similarity using the sum of squared distances (SSD) and finds the optimal number of clusters using Calinski & Harabasz statistics. Then the motifs from the same cluster are marked in the same colour in the online report.
Distinctive features compared with other tools
i-cisTarget is different from other tools in several regards. Only few existing tools provide an enrichment analysis of regulatory data tracks. The few tools that analyse track data, such as the ENCODE ChIP-Seq Significance Tool (12) are limited to TF ChIP-seq tracks only and do not include histone modifications or chromatin accessibility. In addition, to our knowledge no other tool combines track discovery with motif discovery in one single analysis, allowing to compare tracks and motifs, and to combine them into a single set of robust target predictions. Another key difference with other existing tools is the very large motif collection that is used for enrichment analysis, with nearly 10,000 PWMs, collected from various sources and species, clustered and mapped to human/mouse/fly transcription factors either by direct annotation, orthology and/or similarity. i-cisTarget also uses a highly robust statistical analysis––across multiple species––to calculate the feature enrichment. Contrary to most other tools this enrichment is not computed by a hyper-geometric statistical test, but rather by a ranking-and-recovery analysis. Moreover, i-cisTarget offers a unique feature that is the possibility to analyse both genes as well as regions or peaks as input allowing the user to analyse various datasets (e.g. differentially expressed or co-expressed genes; ChIP peaks; differentially active regions; CRM driving similar expression patterns). Also the output of i-cisTarget provides both a list of predicted target regions/enhancers and a list with the closest genes to these target regions. Another interesting feature that we added in the new version is a follow-up tool to compare two analysis results, even between different species, to identify commonly (or distinctively) found motifs that are enriched in both sets. Finally, many small details make i-cisTarget user friendly, such as the selection of the optimal subset of direct target regions from the input; the possibility to combine several motifs and tracks into a single new ranking; the availability to visualize predicted target regions as a custom track in the UCSC genome browser together with the motif and CRM predictions; and the possibility to export a Simple Interaction File (SIF) with motifs/tracks, target regions and the closest genes, that can be imported into Cytoscape (34) to be visualized as a network.
WEB SERVER
We illustrate various types of analyses that can be performed by the i-cisTarget web tool using a case study on Ewing sarcoma (35). Ewing sarcoma is a cancer of bone and soft tissue and is characterized by chromosomal translocations involving EWS gene and transcription factors of the ETS family (ERG, FEV, FLI1, ETV1 and E1AF). EWS-FLI1 fusion product is the most common type as it is found in 90% of the cases (36). Riggi et al. performed ChIP-seq against the chimeric transcription factor EWS-FLI1, as well as ATAC-seq, RNA-seq and ChIP-seq against H3K27ac, all before and after EWS-FLI1 perturbations. Each type of profiling will be used in turn as input to i-cisTarget with the aim of identifying EWS-FLI1 direct target genes, regulatory regions and binding sites.
Analysis of ChIP-seq peaks
i-cisTarget can be used for motif and track discovery on a set of ChIP-seq peaks of a particular transcription factor. Such analysis is usually performed as a quality control step of ChIP-seq data analysis, because the motif of the ChIP'ped transcription factor is expected to be enriched among the binding peaks. In addition, i-cisTarget uses the cross-species motif scores to optimally discriminate between directly bound regions and indirectly bound regions (or false positives). Finally, the motif and track analysis of ChIP-seq peaks can be useful to identify candidate co-factors of the ChIP'ped factor, in case the ChIP'ped regions are enriched for motifs or ChIP-seq peaks of other transcription factors.
As an example we use the ChIP-seq peaks of EWS-FLI1 as input. The authors ChIP'ped the C-terminal portion of FLI1 gene in two Ewing sarcoma cell lines (A673 and SK-N-MC) and defined the 1785 peaks that are present in both cell lines as the ‘core set of EWS-FLI1’ binding sites (provided in the Supplementary Table S1 of the corresponding paper). i-cisTarget analysis on this core set of binding sites identified the motif of the fusion product as the first motif (Figure 2c, i-cisTarget results on the website). Interestingly, ENCODE tracks of PolII ChIP-seq and DNAseI-seq on the Ewing sarcoma cell line SK-N-MC are highly ranked in the results. In addition, the remaining enriched features indicate a vast presence for ETS-family transcription factor motifs and this is indeed in agreement with the authors’ claim that the EWS-FLI1 oncogenic protein displaces ETS-family members from their native binding sites.
Figure 2.
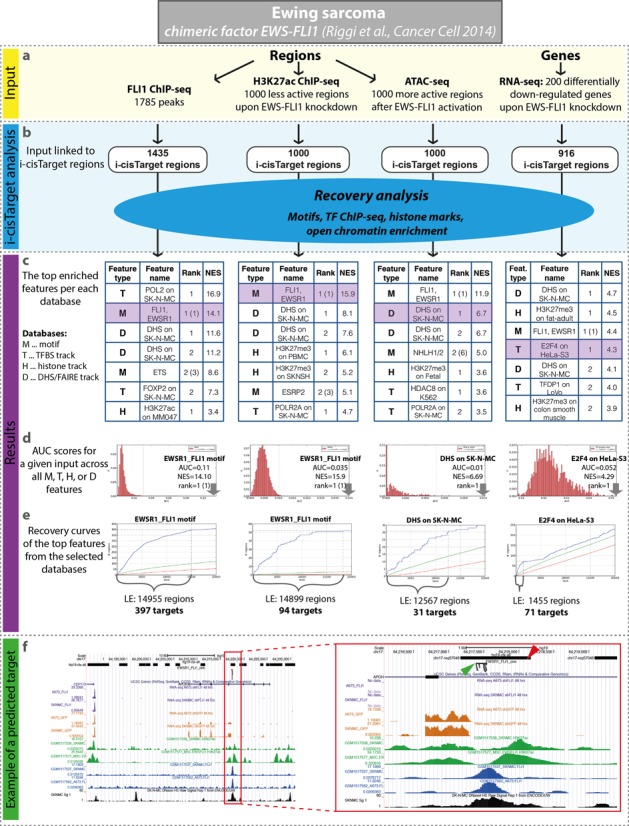
Ewing sarcoma case study. Various i-cisTarget analyses with different types of input, all related to EWS-FLI1 targets in Ewing sarcoma. (a) All input datasets are derived from Riggi et al. (35) and include FLI1 ChIP-seq peaks, top differentially less active peaks based on H3K27ac ChIP-seq upon EWS-FLI1 knockdown, differentially more open regions based on ATAC-seq after EWS-FLI1 activation and differentially downregulated genes after EWS-FLI1 knockdown (see ‘Materials and Methods’ section). (b) Input regions are automatically mapped to CRRs. (c). Each of the sets was analysed independently and reassuringly the expected motif EWS-FLI1 was ranked at the top, alongside regulatory tracks, mainly obtained on the SK-N-MC Ewing sarcoma cell line. Note that the rank of the motifs is represented by two values––the first one for the rank of the cluster of similar motifs, the second one is between brackets and represents the rank of the specific motif. (d) Distributions of AUC scores for a given input across all features in the selected databases (marked in purple in tables (c)) with an arrow indicating the top feature within that database. (e) The recovery curves for the top ranked features within the database, where the leading edge (LE) indicates the number of highly ranked target regions. (f) UCSC genome browser screenshot representing an example of one direct target region (red arrowhead) in the intron of gene APOH, which is included in the set of downregulated genes. This region was predicted as a target of EWS-FLI1 in i-cisTarget analyses, of the top less active H3K37ac peaks and the top 200 downregulated genes as well as FLI1 ChIP-seq peaks. The specific binding site is represented by a cluster of EWS-FLI1 motifs (green arrowhead), which was generated by i-cisTarget subsequent analysis, when the predicted target regions of EWS-FLI1 were scanned for CRMs of this factor. All these tracks are represented on the screenshot (from top to bottom): the CRRs, the predicted cluster of EWS-FLI1 motifs, RNA-seq peaks in SK-N-MC and A673 after shFLI1 (two purple tracks, published in (35)) and control (two orange tracks, published in (35)), H3K27ac peaks in SK-N-MC and MSC cell lines expressing EWS-FLI1 (green tracks, published in (35)), FLI1 ChIP-seq track in SK-N-MC and A673 cell lines (blue tracks, published in (35)) as well as DHS on SK-N-MC which was found as the top track within non-TF regulatory tracks (black track, from ENCODE database (15)).
We also applied i-cisTarget to a previously published benchmark dataset of 36 ChIP-seq datasets selected from FactorBook (37). i-cisTarget identified the correct motif of the ChIP'ped TF for 27 (the motif ranked among the top five motifs, from which for 22 sets are the motifs ranked as the first) of the 36 datasets, when using the top 500 peaks as input. Interestingly, i-cisTarget can also be applied to the entire set of ChIP-peaks (sometimes several thousands of peaks) with the same computational efficiency. As expected, such input is slightly more noisy and yields the correct motif ranked at the first position for 18 of the 36 factors (19 correct motifs ranked among the top five motifs). The results from this analysis are available on the website.
In conclusion, when TF ChIP-seq peaks are analysed, i-cisTarget identifies the motif of the ChIP'ped TF, allowing for quality control, but also for the identification of co-factors and for distinguishing between direct and indirect targets.
Analysis of other regulatory regions
The main advantage of i-cisTarget is the detection of upstream regulators for the regions that are found to be active (or alternatively, repressed), when the binding regulator is not known. To demonstrate this, we analysed the differentially H3K27-acetylated regions upon EWS-FLI1 knockdown in SK-N-MC and A673 cell lines, measured by H3K27ac ChIP-seq. For this, coverage of i-cisTarget regions was quantified using BEDTools (38) and differentially active regions after FLI1 knockdown (hence sites activated by EWS-FLI1) were identified using DESeq2 (39). The top 1000 (ranked on signed-log10(adjusted P-value)) regions that become less active upon EWS-FLI1 knockdown contained the EWS-FLI1 motif as the top ranked motif among all the 9713 motifs, indicating that many of these are directly targeted by EWS-FLI1 (see Figure 2c or results on the i-cisTarget website). We applied the same analysis on ATAC-seq data representing differentially more open regions after EWS-FLI1 activation in mesenchymal stem cells. Again, the top 1000 i-cisTarget regions were found to be enriched with the same EWS-FLI1 motif (see Figure 2c). On the other hand, regions that are activated upon EWS-FLI1 knockdown contained strong enrichment for ETS family factors, especially for ELF1 and GABPA. Such competition was also proposed by Riggi et al. (results available on the i-cisTarget website).
We furthermore applied i-cisTarget to various other sets of regulatory regions, such as similar enhancers that are active in the same cell type, DHS regions of a particular tissue or cell type, p300 bound regions in a tissue and others. The results of these analyses are available as examples on the i-cisTarget website.
Analysis of gene signatures
To demonstrate the use of i-cisTarget on human co-expressed gene sets we re-analysed the RNA-seq data generated with FLI1-knockdown in two EWS cell lines (A673 and SK-N-MC at 48 h). Raw sequence reads were downloaded from SRA, mapped to reference genome (Gencode v18) using TopHat2 (40). Gene expression values were quantified with htseq-count command from HTSeq framework (41) and differential expression analysis was performed using DESeq2 (39) in R/Bioconductor platform. The top 200 significantly (adjusted P-value < 0.05, ranked by log2FC) downregulated genes upon FLI1-knockdown were analysed with i-cisTarget (using AUC threshold = 0.05) and the expected EWS-FLI1 motif was found as the first motif instance while the first ranked DHS track was on SK-N-MC cell line (Figure 2c).
In a larger study we furthermore applied i-cisTarget to 214 TF-perturbation gene signatures for 46 unique TFs (obtained from MSigDB (42)). i-cisTarget identified the correct motif for the perturbed TF for 29 of the 46 TFs and the correct TF ChIP-seq track for 25 of the datasets (for 18 TFs both the motif and ChIP-seq track are found). Note that motif and track enrichment of gene sets can also be performed through another user interface, namely the Cytoscape plugin iRegulon (8) (no web tool), which is based on the same ranking-and-recovery principle. iRegulon is focussed on gene-level analyses, whereby only motif and TF ChIP-seq enrichment analysis can be performed, allowing gene network-reconstruction. The region-based analysis in i-cisTarget on the other hand provides CRM-level analysis, including the detection of enriched histone marks and open chromatin tracks, which are not available in iRegulon and allows detecting specific regulatory target regions, with visualization in the UCSC Genome Browser.
CONCLUSION
The identification of upstream regulators and their direct targets is an important question when analysing regulatory genomics data such as TF binding or open chromatin profiling data. Our tool can be used to address this question in a relatively fast and straightforward way. We demonstrated that i-cisTarget is often able to detect the correct upstream regulators when co-regulated regions are used as input, but also when co-expressed genes are used as input. An important asset of i-cisTarget is the simultaneous analysis of motif enrichment and ‘track enrichment’, being able to combine the best-scoring motifs and tracks to predict regulatory regions underlying the process under study. We believe that identifying enriched regulatory features should become as simple as identifying enriched GO terms in a gene set. As more and more data become available in public databases, it is expected that most types of input data will correlate with one or more earlier performed experiments and i-cisTarget helps to quickly find similarities between the user input data and these available datasets. Since it is based on a generic ranking-and-recovery procedure, this versatile tool can incorporate any genomic data type in the future.
Acknowledgments
The authors thank to ENCODE, Mouse Encode, Roadmap Epigenomics Project and Taipale laboratory for making the datasets publicly available. We thank Rekin's Janky and Dmitry Svetlichnyy for compiling the PWM database.
FUNDING
FWO (www.fwo.be) [G.0704.11N, G.0640.13, G.0791.14 to S.A.]; Special Research Fund (BOF) KU Leuven (http://www.kuleuven.be/research/funding) [PF/10/016, OT/13/103 to S.A.]; HFSP (www.hfsp.org) [RGY0070/2011 to S.A.]; Foundation Against Cancer (http://www.cancer.be) [2012-F2 to S.A.]. IWT PhD fellowship (to H.I.); Kom op Tegen Kanker (Stand up to Cancer), the Flemish cancer society post-doctoral fellowship (to Z.K.A.). Funding for open access charge: IWT.
Conflict of interest statement. None declared.
REFERENCES
- 1.Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48–54. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 3.Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 4.Kwon A.T., Arenillas D.J., Hunt R.W., Wasserman W.W. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda) 2012;2:987–1002. doi: 10.1534/g3.112.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gotea V., Ovcharenko I. DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic Acids Res. 2008;36:W133–W139. doi: 10.1093/nar/gkn300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gotea V., Visel A., Westlund J.M., Nobrega M.A., Pennacchio L.A., Ovcharenko I. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 2010;20:565–577. doi: 10.1101/gr.104471.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janky R., Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Naval Sanchez M., Potier D., et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014;10:e1003731. doi: 10.1371/journal.pcbi.1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas-Chollier M., Herrmann C., Defrance M., Sand O., Thieffry D., van Helden J. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res. 2011;40:e31–e39. doi: 10.1093/nar/gkr1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machanick P., Bailey T.L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu T., Ortiz J.A., Taing L., Meyer C.A., Lee B., Zhang Y., Shin H., Wong S.S., Ma J., Lei Y., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83–R92. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auerbach R.K., Chen B., Butte A.J. Relating genes to function: identifying enriched transcription factors using the ENCODE ChIP-Seq significance tool. Bioinformatics. 2013;29:1922–1924. doi: 10.1093/bioinformatics/btt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst J., Plasterer H.L., Simon I., Bar-Joseph Z. Integrating multiple evidence sources to predict transcription factor binding in the human genome. Genome Res. 2010;20:526–536. doi: 10.1101/gr.096305.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montgomery S.B., Griffith O.L., Sleumer M.C., Bergman C.M., Bilenky M., Pleasance E.D., Prychyna Y., Zhang X., Jones S.J.M. ORegAnno: an open access database and curation system for literature-derived promoters, transcription factor binding sites and regulatory variation. Bioinformatics. 2006;22:637–640. doi: 10.1093/bioinformatics/btk027. [DOI] [PubMed] [Google Scholar]
- 18.Pennacchio L.A., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 19.Ferretti V., Poitras C., Bergeron D., Coulombe B., Robert F., Blanchette M. PReMod: a database of genome-wide mammalian cis-regulatory module predictions. Nucleic Acids Res. 2007;35:D122–D126. doi: 10.1093/nar/gkl879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimitrieva S., Bucher P. UCNEbase—a database of ultraconserved non-coding elements and genomic regulatory blocks. Nucleic Acids Res. 2013;41:D101–D109. doi: 10.1093/nar/gks1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouse ENCODE Consortium C., Stamatoyannopoulos J.A., Snyder M., Hardison R., Ren B., Gingeras T., Gilbert D.M., Groudine M., Bender M., Kaul R., et al. An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13:418–422. doi: 10.1186/gb-2012-13-8-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer L.R., Zweig A.S., Hinrichs A.S., Karolchik D., Kuhn R.M., Wong M., Sloan C.A., Rosenbloom K.R., Roe G., Rhead B., et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herrmann C., de Sande B.V., Potier D., Aerts S. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012;40:e114. doi: 10.1093/nar/gks543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frith M.C., Li M.C., Weng Z. Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aerts S., Lambrechts D., Maity S., Van Loo P., Coessens B., De Smet F., Tranchevent L.-C., De Moor B., Marynen P., Hassan B., et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 27.Stuart J.M., Segal E., Koller D., Kim S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 28.Roadmap Epigenomics Consortium. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan J., Enge M., Whitington T., Dave K., Liu J., Sur I., Schmierer B., Jolma A., Kivioja T., Taipale M., et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154:801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 30.Verfaillie A., Imrichova H., Atak Z.K., Dewaele M., Rambow F., Hulselmans G., Christiaens V., Svetlichnyy D., Luciani F., Van den Mooter L., et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nature. 2015;6:6683–6698. doi: 10.1038/ncomms7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Portales-Casamar E., Thongjuea S., Kwon A.T., Arenillas D., Zhao X., Valen E., Yusuf D., Lenhard B., Wasserman W.W., Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–D110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahony S., Auron P.E., Benos P.V. DNA familial binding profiles made easy: comparison of various motif alignment and clustering strategies. PLoS Comput. Biol. 2007;3:e61. doi: 10.1371/journal.pcbi.0030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smoot M.E., Ono K., Ruscheinski J., Wang P.-L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riggi N., Knoechel B., Gillespie S.M., Rheinbay E., Boulay G., Suvà M.L., Rossetti N.E., Boonseng W.E., Oksuz O., Cook E.B., et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. 2014;26:668–681. doi: 10.1016/j.ccell.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aman P. Fusion genes in solid tumors. Semin. Cancer Biol. 1999;9:303–318. doi: 10.1006/scbi.1999.0130. [DOI] [PubMed] [Google Scholar]
- 37.Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550–570. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36–R48. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]