


Abstract
ProtPhylo is a web-based tool to identify proteins that are functionally linked to either a phenotype or a protein of interest based on co-evolution. ProtPhylo infers functional associations by comparing protein phylogenetic profiles (co-occurrence patterns of orthology relationships) for more than 9.7 million non-redundant protein sequences from all three domains of life. Users can query any of 2048 fully sequenced organisms, including 1678 bacteria, 255 eukaryotes and 115 archaea. In addition, they can tailor ProtPhylo to a particular kind of biological question by choosing among four main orthology inference methods based either on pair-wise sequence comparisons (One-way Best Hits and Best Reciprocal Hits) or clustering of orthologous proteins across multiple species (OrthoMCL and eggNOG). Next, ProtPhylo ranks phylogenetic neighbors of query proteins or phenotypic properties using the Hamming distance as a measure of similarity between pairs of phylogenetic profiles. Candidate hits can be easily and flexibly prioritized by complementary clues on subcellular localization, known protein–protein interactions, membrane spanning regions and protein domains. The resulting protein list can be quickly exported into a csv text file for further analyses. ProtPhylo is freely available at http://www.protphylo.org.
INTRODUCTION
Advances in sequencing technologies and genome annotation tools continuously increase the repertoire of protein-coding genes in numerous organisms. The number of sequenced genomes is growing exponentially, with over 10 000 prokaryotic and eukaryotic species sequenced to date (1). However, with the exception of well-studied model organisms, the majority of species-specific protein sets remains functionally uncharacterized even though high-throughput functional annotation projects have been initialized (2). One of the most direct approaches to understand the function of a protein of interest consists of elucidating its interaction partners. Functional links can be obtained for example by experimental analysis of physical protein–protein interactions (e.g. protein complex purification) or gene–gene relationships (e.g. double mutants phenotyping and correlated gene expression). However, genome-wide surveys of functional associations remain experimentally challenging in many organisms (3).
In silico-based predictions of functionally linked proteins often allow inferring a function for uncharacterized components via 'guilt-by-association' with known components (4,5). One such method is based on phylogenetic profiling (6), whose predictive power increases as more sequenced genomes from diverse taxonomic groups become available (7). Phylogenetic profiling predicts functional associations on the assumption that if proteins co-occur, despite multiple evolutionary events of speciation, gene loss and lateral transfer across a large number of genomes, then they are functionally coupled. The first step involves the identification of orthologs for a protein of interest in several genomes, defining what is called a 'protein phylogenetic profile'. Next is the search for proteins, within the same genome, that show a correlated pattern of presence and absence. Many successful case studies support the application of phylogenetic profiling to identify novel components of a biological process (e.g. a biochemical pathway or a multi-subunit protein complex) (8,9), to annotate orphan proteins (10,11) and to discover proteins underlying a phenotype of interest (12,13).
Here we present ProtPhylo (www.protphylo.org), a web-based tool for prediction of protein-to-protein and phenotype-to-protein functional associations based on phylogenetic profiling. ProtPhylo achieves flexibility and state-of-the-art taxonomic and functional coverage by generating phylogenetic profiles for 9.7 million non-redundant protein sequences across 2048 organisms and by implementing four independent orthology detection algorithms. In addition, it provides an integrated framework for fast, easy and flexible prioritization of phylogenetic neighbors based on widely used tools for prediction of subcellular localization (14–18), protein domains (19), membrane spanning regions (18,20) and complementary evidence of protein–protein interactions (21).
THE PROTPHYLO PIPELINE
Genome selection
The first step in the ProtPhylo pipeline is the selection of relevant genomes for phylogenetic profiling (Figure 1). As in EggNOGv.4 (22), we restricted our analysis to publicly available and high-quality genome datasets as for genomic completeness, sequencing coverage and accuracy of genome annotation. While such criteria help minimizing false orthology assignment (23,24), the inclusion of species from multiple taxonomic levels at different evolutionary distances is key to maximize the resolution of coupled evolutionary patterns (7). The resulting species set covers a total of 2048 organisms, including 1678 bacteria, 115 archaea and 255 eukaryotes. The species list can be downloaded directly from the ProtPhylo web server.
Figure 1.
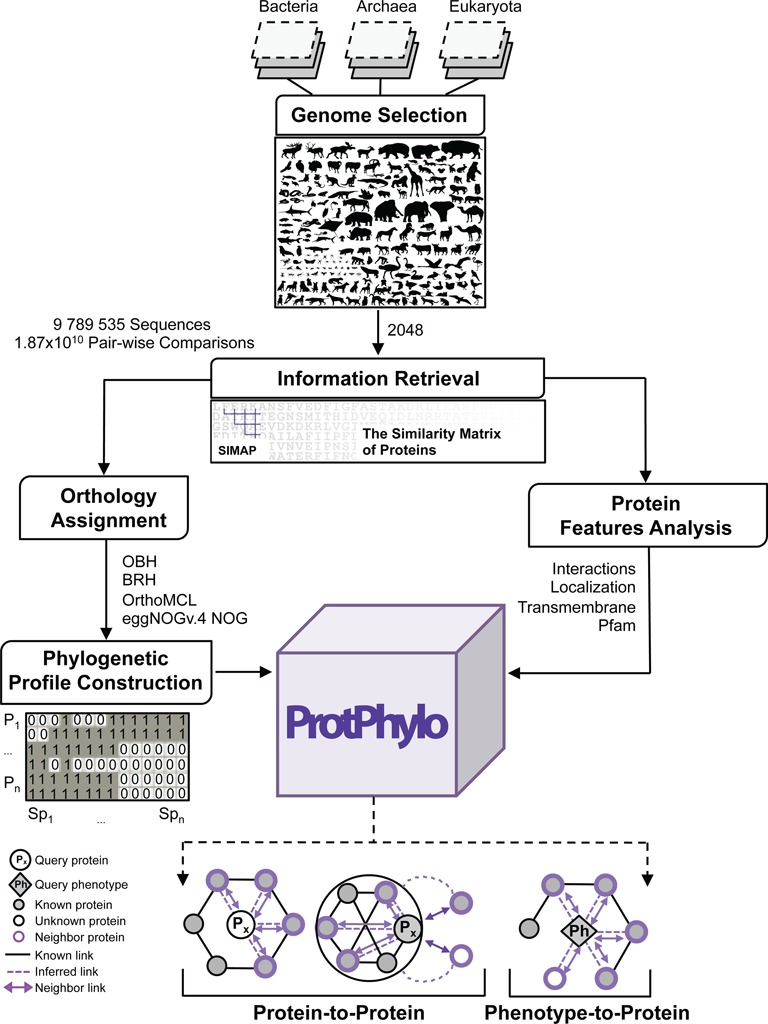
Schematic representation of the ProtPhylo Pipeline.
Retrieval of sequence similarity scores
For all 2048 organisms, non-redundant protein sequences were retrieved from the Similarity Matrix of Proteins (SIMAP) database (25) and filtered by sequence length (>10 amino acids) and quality (<20% non-standard amino acids). The corresponding 9 789 535 protein sequences represent a nearly even sampling of prokaryotic and eukaryotic proteomes (Table 1). Roughly, 86% of all sequences could be annotated based on protein IDs mapping between the original sequence repository (ENSEMBL, NCBI RefSeq, JGI) and Uniprot database. Sequence annotations for Saccharomyces cerevisiae, Caenorhabditis elegans and Danio rerio were based on the Saccharomyces Genome Database, Wormbase and ZFIN database, respectively. Sequence similarity scores for 1.87 x 1010 pair-wise comparisons were generated by the SIMAP initiative as described in EggNOGv.4 (22). Briefly, sequence alignments and similarity scores were generated with the FASTA algorithm and then recalculated using the basic local alignment search tool (BLAST) with compositional adjustment of the amino acid substitution matrix and bit score cutoff ≥50.
Table 1. Source and number of non-redundant sequences for 2048 organisms included in ProtPhylo as of February 2015.
| Kingdom (# species) | Non-redundant sequences | Database |
|---|---|---|
| Bacteria (1678) | 5 559 635 | NCBI RefSeq |
| Archaea (115) | 256 635 | NCBI RefSeq |
| Eukaryota (255) | 3 973 265 | ENSEMBL, Uniprot, |
| NCBI RefSeq, JGI |
Orthology assignment and construction of phylogenetic profiles
Accurate orthology prediction is a crucial step for the construction of protein phylogenetic profiles. An overwhelming number of alternative methodologies exist for genome-wide orthology inference (26) and several attempts have already been made to compare their relative performance (27,28). However, it remains challenging to draw a conclusion on which method is the best. Instead, the choice of an orthology detection method over others largely depends on the kind of functional conservation being predicted (e.g. co-expression, molecular function, involvement in similar pathways, protein–protein interaction, etc.) as well as on the number, diversity and evolutionary distances of the species being compared (26–28). Currently, ProtPhylo implements four commonly used, BLAST-based algorithms for orthology assignments (Figure 2A): One-way Best-Hits (OBH), Best-Reciprocal-Hits (BRH) (29), OrthoMCL (30) and eggNOGv.4 (22). OBH and BRH rely on pair-wise sequence comparisons and determine orthology using a simple BLAST cut-off criterion, (E-value < 10−5). Instead, OrthoMCL and eggNOGv.4 extend the sequence similarity search over multiple proteomes at once to generate groups of orthologous proteins. Both, OrthoMCL and eggNOGv.4 algorithms, derive orthologous groups based on BRH (E-value < 10−5) as their first step but apply different clustering techniques to assemble protein groups from multiple species, Markov Cluster (MCL) (30) and triangular linkage (31), respectively. In addition to the default parameters used in OrthoMCL, ProtPhylo derives orthologous groups also based on different settings for percent match length (≥0 or ≥50%) and inflation index (1.1, 1.5 or 5). The first refers to the percentage of positive-scoring matches of the high-scoring pairs and the second corresponds to the parameter used by MCL to define the tightness of orthologous groups. Here, the higher the inflation indexes, the tighter the size of the orthologous groups (Figure 2B).
Figure 2.
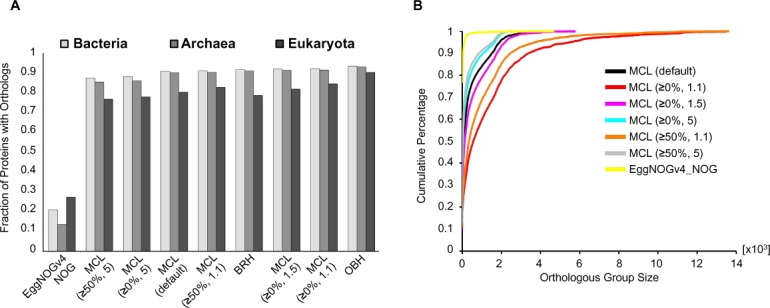
Differences in orthology prediction methods. (A) Percentage of proteins in each kingdom having at least one orthologous protein in any other species. (B) Cumulative percentage of proteins for different orthologous group size. The smaller the inflation index or the percent match length, the larger the orthologous group size. MCL (≥x, y) refers to the OrthoMCL algorithm with x representing the percent match length and y representing the inflation index.
Next, for all 9.7 million proteins we generated phylogenetic profiles across 2048 organisms using the abovementioned orthology inference methods. Here, the phylogenetic profile of a protein or a phenotype is represented by a binary string with n entries, where n corresponds to the size of the species set and the entry indicates the presence (1) or absence (0) of an ortholog or a similar phenotype across species.
Retrieval of protein features
ProtPhylo includes functional annotations for all 9.7 million proteins based on evidence of subcellular localization (14–18), presence of transmembrane helices (18,20), protein domain families (19) and complementary evidence of protein–protein interactions (21). Subcellular localization is either predicted by applying sequence-based computational strategies, as in TargetP 1.1 (15), MitoProt II (16), and LocTree3 (14), or retrieved by Uniprot database. Briefly, TargetP 1.1 uses N-terminal sequence information to discriminate between proteins targeted to the mitochondrion, the chloroplast, the secretory pathway and ‘others’. MitoProt II computes the probability (MitoProt II Score) that a protein has a mitochondrial-targeting sequence. Prediction of mitochondrial localization for Homo sapiens and Mus musculus is also retrieved from MitoCarta (17). LocTree3 predicts protein localization to at least twelve of the major subcellular locations of prokaryotic and eukaryotic cells (nucleus, cytoplasm, mitochondrion, plasma membrane, Golgi apparatus, endoplasmic reticulum, vacuole, peroxisome, plastid, chloroplast, extracellular region and fimbrium). Currently, LocTree3 predictions are available for ∼1500 species in ProtPhylo (68% of protein sequences). Protein transmembrane helices are predicted by using TMHMM v2.0 algorithm (20) as well as annotated from Uniprot database based on experimental and computational evidence. Protein domain families are predicted by hmmscan program against Pfam-A domains (Pfam 27.0) (19).
PROTPHYLO WEB SERVER IMPLEMENTATION
Input
Users can search for proteins, within a query organism, that co-evolve with either a phenotype (Phenotype Phylogenetic Profiling), for example ion uptake, thermogenesis, or multicellularity, or a protein of interest (Protein Phylogenetic Profiling) and are therefore likely to be functionally associated. Phenotype Phylogenetic Profiling can only be applied when users have a priori knowledge of the presence and/or the absence of the phenotype across any of the 2048 organisms in ProtPhylo (Species WITH and/or Species WITHOUT the Phenotype of Interest). In both search options, users can select among four main orthology methods (OBH, BRH, OrthoMCL and eggNOGv.4), as well as five additional OrthoMCL settings (OBH is set as the default orthology method). ProtPhylo uses all 2048 organisms to generate a protein phylogenetic profile, while the set of species used to generate a phenotype phylogenetic profile can be defined by the user.
Output
ProtPhylo compares the query phylogenetic profiles (protein or phenotype) to the profiles of all other proteins within the query organism and ranks proteins based on their similarity scores, from the smallest (closest phylogenetic neighbor) to the largest. Here, we use the Hamming distance (HD) to quantify the similarity between pairs of phylogenetic profiles (6). This corresponds to the number of positions whereby the two binary vectors have different entries. In total, ProtPhylo takes ∼2 s of calculation time for each query, when default settings are used. In addition to the HD, each row of the output list (phylogenetic neighbor) contains information on protein IDs and names from relevant protein repositories. Further protein details can be found through hyperlinks (Source Protein ID). When selecting OrthoMCL or eggNOGv.4 as orthology detection methods, users can also retrieve the list of orthologs for each ranked protein (Orthologs). For OBH and BRH, the retrieval of orthologs is available for the following query organisms: H. sapiens, M. musculus, S. cerevisiae, C. elegans, D. melanogaster and D. rerio. The output of a Protein Phylogenetic Profiling search includes two additional calculations: the HD Percentile and the Reciprocal HD Percentile. The former refers to the percentage of proteins within the query proteome that have equal or lower HD than the phylogenetic neighbor HD, while the latter is calculated when clicking the magnifier icon and reflects the HD percentile of the user-defined query protein if the phylogenetic neighbor is used as a query. By default, ProtPhylo reports a protein list with less than or equal to the fifth percentile. The output list can be further prioritized directly in ProtPhylo based on five complementary filtering criteria: cut-off HD values and percentile; combined (And) or stand-alone (Or) evidence of subcellular localization; presence (>0) or absence (=0) of transmembrane helices; presence of conserved Pfam domains (by Pfam ID or name); keywords (gene symbol, protein IDs and names); confidence score for functional associations predicted by STRING (21), (STRING score).
Phenotype-to-protein: the MCU case study
As an illustrative example of Phenotype Phylogenetic Profiling, we used ProtPhylo web interface to predict human proteins that co-evolve with the ability of mitochondria to uptake calcium via a uniport mechanism (32,33). The underlying machinery, called ‘the mitochondrial calcium uniporter’ (MCU), consists of a highly selective calcium channel with unique biophysical properties, being high-capacity, dependent on membrane potential, calcium-selective even at low concentration of free cytosolic calcium and sensitive to nanomolar concentrations of ruthenium derivatives (34). Despite intense efforts dating back to over 60 years ago, the molecular nature of MCU has evaded traditional biochemical strategies as well as genome-wide RNAi screens. Recently, this mystery was uncovered by searching for human mitochondrial proteins that are conserved in vertebrates and kinetoplastids but not in yeast, which is unable to perform calcium uptake (12), (Figure 3A). Here, phylogenetic profiling was applied to predict phenotype-to-protein functional associations, leading to the discovery of uniporter's regulatory and structural subunits, Micu1-2 and Mcu, respectively (12,13). As shown in Figure 3B, ProtPhylo predicts 35 human proteins as having the same phylogenetic profile of the phenotype of interest (HD = 0) across the six taxa and being localized to mitochondria (combined evidence from MitoProt II score, TargetP 1.1, Loctree3, and Uniprot). Candidate proteins include Micu1-2 and Mcu, as well as other components of the uniporter protein complex such as Mcub (35).
Figure 3.

Example of phenotype-to-protein functional associations predicted by Phenotype Phylogenetic Profiling in ProtPhylo. (A) Mitochondria calcium uptake (Phenotype) is common to vertebrate and protozoa, yet not measurable in Saccharomyces cerevisiae (Phylogenetic Profile). Human proteins of the mitochondrial calcium uptake channel were predicted by looking for mitochondria-localized proteins that have the same phylogenetic profile of the calcium uptake phenotype (12) and have predicted transmembrane domains (13). (B) The ProtPhylo web interface query for the phenotype described in (A). Known components of the human (query organism) mitochondrial calcium uniporter are found within the 35 phylogenetic neighbors predicted by ProtPhylo.
Protein-to-protein: the Stim1 case study
As an illustrative example of Protein Phylogenetic Profiling, we used ProtPhylo to predict proteins that are functionally linked to Stim1, stromal interaction molecule 1. Stim1 is a key signaling protein regulating the influx of calcium through the plasma membrane in response to InsP3-induced depletion of the endoplasmic reticulum calcium pool (36). Calcium entry through the plasma membrane occurs through Calcium Release-Activated Calcium (CRAC) channels whose molecular identity remained unknown till 2006. After the discovery of Stim1 in 2005 by genome-wide RNAi screening efforts (37,38), it followed the identification of the CRAC channel subunit, Orai1, through three other genome-wide RNAi screens (39–41). Here, we asked whether ProtPhylo could predict the functional association between Stim1 and Orai1 solely based on phylogenetic profiling and protein-feature analysis. As shown in Figure 4, we searched for human proteins that co-occur with Stim1 across all 2048 organisms (≤1st HD percentile), localize to the plasma membrane (LocTree3, Uniprot) but not to the mitochondria (MitoCarta) and have at least one transmembrane domain. As a result, ProtPhylo identifies 16 proteins matching the selected criteria, with Orai1 being the top phylogenetic neighbor of Stim1 (HD = 5).
Figure 4.
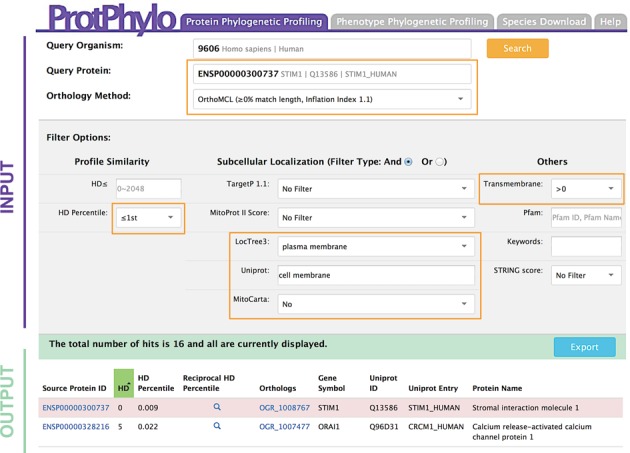
Example of Protein Phylogenetic Profiling. The ProtPhylo web interface query used to predict human proteins (query organism) functionally associated with Stim1 (query protein) is shown. Here, OrthoMCL (≥0% match length, inflation index 1.1) is used as orthology detection method. Known components of the CRAC channel represent the top phylogenetic neighbors (smallest HD).
DISCUSSION
ProtPhylo aims to provide a fast, flexible and user-friendly tool to assist biologists seeking functional clues for a protein or a phenotype of interest. Several features distinguish ProtPhylo from other web servers (21,30,42–43). First, it implements different orthology detection methods to generate phylogenetic profiles. Second, ProtPhylo can operate on two types of inputs, protein-based and phenotype-based phylogenetic profiles. Third, it covers three domains of life and retrieves functional associations for proteins from any of 2048 organisms. To emphasize its suitability as a discovery tool, we validated its performance with datasets of known human protein complexes (CORUM, (44)), cellular components from the Gene Ontology (GO) database (45) and metabolic and signaling pathways from KEGG (46). Overall, we find that ProtPhylo reaches the highest protein pairs recall rate when applied to CORUM dataset of 1736 manually curated human protein complexes (data not shown). Varying the orthology method for phylogenetic analysis in ProtPhylo shows little effect on protein pairs recall rates. However, when combining all orthology methods, the recall rate increases for all three datasets, indicating that different orthology methods recall a different subset of true positive interactions, as also shown in Figure 5. This highlights the value of including more than one orthology method in ProtPhylo, a unique feature that distinguishes ProtPhylo from other web servers (21,30,42–43). Therefore, it could be advantageous to run ProtPhylo with different methods or use them in combination to increase the rate of true positive predictions, while using other filtering options available in ProtPhylo to decrease the rate of false positives. In summary, ProtPhylo web server offers users the possibility to narrow down the number of testable hypotheses through the extension of phylogenetic profiling and comparative biology analyses to an ever growing sequence space.
Figure 5.
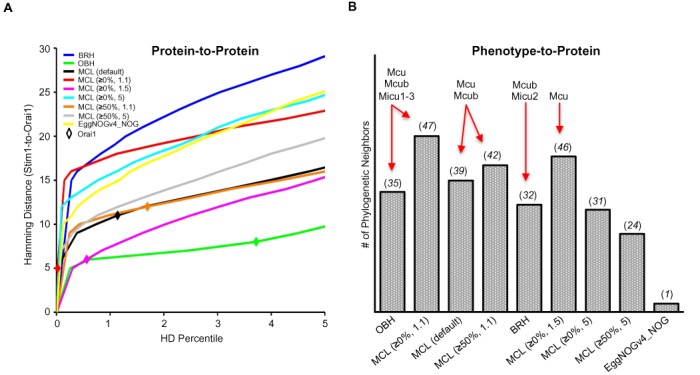
Effect of different orthology detection methods on phylogenetic profiling. (A) Hamming distance between human Stim1 (query protein) and Orai1 (phylogenetic neighbor) when using different orthology methods and parameters. The strongest functional association (lower HD and HD Percentile) can be predicted using OrthoMCL (≥0% match length, inflation index 1.1). (B) Number of phylogenetic neighbor proteins predicted to co-evolve with the calcium uptake phenotype (HD = 0) based on different orthology detection methods. The presence of known components of the mitochondrial calcium uptake channel is shown.
Acknowledgments
Thanks to Thomas Rattei and the SIMAP initiative for providing the similarity matrix data and the LocTree3 team for providing subcellular localization data. Thanks to Johannes Soeding, Julien Gagneur and Martin Jastroch for helpful discussions and to Helmut Blum, Alexander Graf and Max Grimm for technical assistance.
FUNDING
German Research Foundation (DFG) under the Emmy Noether Programme [PE 2053/1-1 to F.P.]; Bavarian Ministry of Sciences, Research and the Arts in the framework of the Bavarian Molecular Biosystems Research Network [D2-F5121.2-10c/4822 to Y.C.]. Funding for open access charge: German Research Foundation (DFG) under the Emmy Noether Programme [PE 2053/1-1].
Conflict of interest statement. None declared.
REFERENCES
- 1.Reddy T.B., Thomas A.D., Stamatis D., Bertsch J., Isbandi M., Jansson J., Mallajosyula J., Pagani I., Lobos E.A., Kyrpides N.C. The Genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res. 2015;43:D1099–D1106. doi: 10.1093/nar/gku950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koskinen P., Toronen P., Nokso-Koivisto J., Holm L. PANNZER: high-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics. 2015:btu851. doi: 10.1093/bioinformatics/btu851. [DOI] [PubMed] [Google Scholar]
- 3.Radivojac P., Clark W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A., et al. A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013;10:221–227. doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N., et al. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perocchi F., Jensen L.J., Gagneur J., Ahting U., von Mering C., Bork P., Prokisch H., Steinmetz L.M. Assessing systems properties of yeast mitochondria through an interaction map of the organelle. PLoS Genet. 2006;2:e170. doi: 10.1371/journal.pgen.0020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pellegrini M., Marcotte E.M., Thompson M.J., Eisenberg D., Yeates T.O. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skunca N., Dessimoz C. Phylogenetic profiling: how much input data is enough? PloS One. 2015;10:e0114701. doi: 10.1371/journal.pone.0114701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabaldon T., Rainey D., Huynen M.A. Tracing the evolution of a large protein complex in the eukaryotes, NADH: ubiquinone oxidoreductase (Complex I) J. Mol. Biol. 2005;348:857–870. doi: 10.1016/j.jmb.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 9.Gertz J., Elfond G., Shustrova A., Weisinger M., Pellegrini M., Cokus S., Rothschild B. Inferring protein interactions from phylogenetic distance matrices. Bioinformatics. 2003;19:2039–2045. doi: 10.1093/bioinformatics/btg278. [DOI] [PubMed] [Google Scholar]
- 10.Huynen M.A., Snel B., Bork P., Gibson T.J. The phylogenetic distribution of frataxin indicates a role in iron-sulfur cluster protein assembly. Hum. Mol. Genet. 2001;10:2463–2468. doi: 10.1093/hmg/10.21.2463. [DOI] [PubMed] [Google Scholar]
- 11.Baughman J.M., Perocchi F., Girgis H.S., Plovanich M., Belcher-Timme C.A., Sancak Y., Bao X.R., Strittmatter L., Goldberger O., Bogorad R.L., et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perocchi F., Gohil V.M., Girgis H.S., Bao X.R., McCombs J.E., Palmer A.E., Mootha V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Stefani D., Raffaello A., Teardo E., Szabo I., Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldberg T., Hecht M., Hamp T., Karl T., Yachdav G., Ahmed N., Altermann U., Angerer P., Ansorge S., Balasz K., et al. LocTree3 prediction of localization. Nucleic Acids Res. 2014;42:W350–W355. doi: 10.1093/nar/gku396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emanuelsson O., Nielsen H., Brunak S., von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 16.Claros M.G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 17.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K., et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magrane M., Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R., Heger A., Hetherington K., Holm L., Mistry J., et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 21.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell S., Forslund K., Szklarczyk D., Trachana K., Roth A., Huerta-Cepas J., Gabaldon T., Rattei T., Creevey C., Kuhn M., et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014;42:D231–D239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trachana K., Larsson T.A., Powell S., Chen W.H., Doerks T., Muller J., Bork P. Orthology prediction methods: a quality assessment using curated protein families. Bioessays. 2011;33:769–780. doi: 10.1002/bies.201100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnhammer E.L., Gabaldon T., Sousa da Silva A.W., Martin M., Robinson-Rechavi M., Boeckmann B., Thomas P.D., Dessimoz C. Big data and other challenges in the quest for orthologs. Bioinformatics. 2014;30:2993–2998. doi: 10.1093/bioinformatics/btu492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rattei T., Tischler P., Gotz S., Jehl M.A., Hoser J., Arnold R., Conesa A., Mewes H.W. SIMAP–a comprehensive database of pre-calculated protein sequence similarities, domains, annotations and clusters. Nucleic Acids Res. 2010;38:D223–D226. doi: 10.1093/nar/gkp949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzniar A., van Ham R.C., Pongor S., Leunissen J.A. The quest for orthologs: finding the corresponding gene across genomes. Trends Genet. 2008;24:539–551. doi: 10.1016/j.tig.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Hulsen T., Huynen M.A., de Vlieg J., Groenen P.M. Benchmarking ortholog identification methods using functional genomics data. Genome Biol. 2006;7:R31. doi: 10.1186/gb-2006-7-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen F., Mackey A.J., Vermunt J.K., Roos D.S. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PloS One. 2007;2:e383. doi: 10.1371/journal.pone.0000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huynen M.A., Bork P. Measuring genome evolution. Proc. Natl. Acad. Sci. U.S.A. 1998;95:5849–5856. doi: 10.1073/pnas.95.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L., Stoeckert C.J. Jr, Roos D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller J., Szklarczyk D., Julien P., Letunic I., Roth A., Kuhn M., Powell S., von Mering C., Doerks T., Jensen L.J., et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010;38:D190–D195. doi: 10.1093/nar/gkp951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deluca H.F., Engstrom G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. U.S.A. 1961;47:1744–1750. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vasington F.D., Murphy J.V. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 1962;237:2670–2677. [PubMed] [Google Scholar]
- 34.Kirichok Y., Krapivinsky G., Clapham D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 35.Raffaello A., De Stefani D., Sabbadin D., Teardo E., Merli G., Picard A., Checchetto V., Moro S., Szabo I., Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cahalan M.D. STIMulating store-operated Ca(2+) entry. Nat. Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roos J., DiGregorio P.J., Yeromin A.V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J.A., Wagner S.L., Cahalan M.D., et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liou J., Kim M.L., Heo W.D., Jones J.T., Myers J.W., Ferrell J.E. Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vig M., Peinelt C., Beck A., Koomoa D.L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang S.L., Yeromin A.V., Zhang X.H., Yu Y., Safrina O., Penna A., Roos J., Stauderman K.A., Cahalan M.D. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S.H., Tanasa B., Hogan P.G., Lewis R.S., Daly M., Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 42.Dittmar W.J., McIver L., Michalak P., Garner H.R., Valdez G. EvoCor: a platform for predicting functionally related genes using phylogenetic and expression profiles. Nucleic Acids Res. 2014;42:W72–W75. doi: 10.1093/nar/gku442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hulsen T., Groenen P.M., de Vlieg J., Alkema W. PhyloPat: an updated version of the phylogenetic pattern database contains gene neighborhood. Nucleic Acids Res. 2009;37:D731–D737. doi: 10.1093/nar/gkn645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruepp A., Waegele B., Lechner M., Brauner B., Dunger-Kaltenbach I., Fobo G., Frishman G., Montrone C., Mewes H.W. CORUM: the comprehensive resource of mammalian protein complexes–2009. Nucleic Acids Res. 2010;38:D497–D501. doi: 10.1093/nar/gkp914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gene Ontology, C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]