


Abstract
JPred4 (http://www.compbio.dundee.ac.uk/jpred4) is the latest version of the popular JPred protein secondary structure prediction server which provides predictions by the JNet algorithm, one of the most accurate methods for secondary structure prediction. In addition to protein secondary structure, JPred also makes predictions of solvent accessibility and coiled-coil regions. The JPred service runs up to 94 000 jobs per month and has carried out over 1.5 million predictions in total for users in 179 countries. The JPred4 web server has been re-implemented in the Bootstrap framework and JavaScript to improve its design, usability and accessibility from mobile devices. JPred4 features higher accuracy, with a blind three-state (α-helix, β-strand and coil) secondary structure prediction accuracy of 82.0% while solvent accessibility prediction accuracy has been raised to 90% for residues <5% accessible. Reporting of results is enhanced both on the website and through the optional email summaries and batch submission results. Predictions are now presented in SVG format with options to view full multiple sequence alignments with and without gaps and insertions. Finally, the help-pages have been updated and tool-tips added as well as step-by-step tutorials.
INTRODUCTION
Knowledge of a protein's three-dimensional structure is central to understanding the protein's detailed function. Although recent developments in structural biology (1–4) have led to an acceleration in the rate of three-dimensional structure determination by X-ray crystallography, nuclear magnetic resonance and 3D-EM techniques, in January 2015 there were still just 105 732 protein structures known (http://www.ebi.ac.uk/pdbe) (5) compared to almost 90 million sequences (http://www.ebi.ac.uk/uniprot/TrEMBLstats) (6). The routine use of massively parallel DNA sequencing technologies today means knowledge of protein sequences will continue to outpace structural biology for the foreseeable future. As a consequence, there is a need for accurate methods to predict structural and functional features from the amino acid sequence. Over the last 30 years, techniques to predict the three-state secondary structure of the protein (α-helix, β-strand and coil: i.e. all other states) have increased in accuracy from around 50% in 1983 (7) to over 80% today (8–11) which is close to the estimated maximum for prediction from multiple alignment (12). Although knowledge of the secondary structure alone is not as useful as a full three-dimensional model, secondary structure predictions provide important constraints for fold-recognition techniques (13–17) as well as in homology modelling (18,19), ab initio (20–24) and constraint-based tertiary structure prediction methods (25–27). Secondary structure predictions can also help in the identification of functional domains and may be used to guide the rational design of site-specific or deletion mutation experiments.
Although hundreds of papers have been published describing methods for protein secondary structure prediction, three of the most widely used are JPred, PSIPRED and PredictProtein. JPred (v. 3.0) (11) gave 81.5% three-state accuracy (Q3), PSIPRED v.3.0 (28) reported accuracy of 81.4%, while the current PSIPRED V 3.2 server, which includes a broad suite of prediction algorithms, quotes 81.6%. (http://bioinf.cs.ucl.ac.uk/psipred). There is no recent blind prediction test for the PROFphd secondary structure prediction algorithm in the PredictProtein (29) secondary structure prediction method, though the earlier PROFsec reported 76% (30).
In this paper we summarize the current performance and features of the upgraded JPred server (JPred4) which incorporates the secondary structure and solvent accessibility prediction program JNet v.2.3.1.
MATERIALS AND METHODS
The basic usage pattern for JPred4 is the same as for JPred3 (11). The user can submit a single protein sequence, a multiple sequence alignment (MSA) or a batch of single protein sequences for prediction. Results are returned either interactively through a web page or as a summary email that directs the user to results on the JPred4 website.
The look and feel of the JPred4 web server has been changed significantly compared to JPred3 by embracing contemporary web technologies, the Bootstrap framework (www.getbootstrap.com/) and custom JavaScript. These changes allow smoother user interaction through the use of ‘tooltips’ that pop up to present help on each option in an easy-to-read form without the need to leave the page. The Bootstrap framework provides a modern look and feel to the website as well as improving usability on devices such as tablets and phones with different screen sizes and resolutions. Figure 1 illustrates the appearance of the advanced submission page showing the use of tooltips to get help about each option. As well as updates to the help pages, step-by-step tutorials with screenshots are a new addition that helps users to obtain maximum benefit from the JPred4 server.
Figure 1.
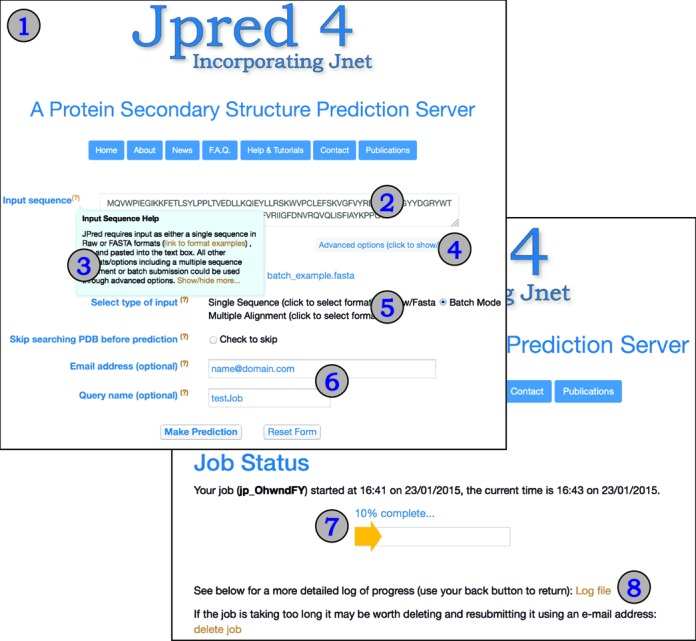
(1) Screenshot of the JPred4 job submission page with single sequence submission field (2) and an example of a tool-tip message (3). Advanced options are opened on request (4) and include input file upload, format selection (5) as well as optional email and query name fields (6). (7) Job progress page with access to the detailed job run log file (8).
Prediction algorithm
As with JPred3, JPred4 makes secondary structure and residue solvent accessibility predictions by the JNet algorithm (11,31). However, in JPred4, the JNet 2.0 neural network-based predictor has been retrained to make JNet 2.3.1 by 7-fold cross-validation using one representative for each of the 1358 SCOPe/ASTRAL v.2.04 superfamily domain sequences (32). Multiple alignments for each sequence were built by PSI-BLAST (33) through searching UniRef90 v.2014_07 (34). In addition to retraining, the HMM building step in JNet was updated to HMMer 3 (35) and some improvements were made to the code to simplify management and future algorithmic developments. The final accuracy of JNet 2.3.1 was assessed in a blind test on 150 sequences from 150 superfamilies not used in training. The 150 superfamily sequences were selected to reproduce a similar distribution of secondary structure compositions as the training structures in order to avoid biasing the reported accuracy of the blind test results. On the blind test, the average secondary structure prediction Q3 score increased to 82.0% from 81.5% for JNet v.2.0, and solvent accessibility prediction accuracy rose to 90.0, 83.6 and 78.1% from 88.9, 82.4 and 77.8% for JNet v.2.0 for each of >0, >5 and >25% relative solvent accessibility thresholds.
JPred4 results reporting
JPred3 has been widely used in teaching and integrated into many bioinformatics pipelines across the world. Accordingly, in order to maintain support for legacy courses and scripts, the results options in JPred4 include all the original formats and styles (PDF, HTML, etc.) as well as the intermediary processing files. In addition to these outputs, JPred4 reports have been enhanced to include more visualization options and to present a complete picture of the alignment generated for prediction including all insertions.
Figure 2 summarizes the main results page while Figure 3 shows examples of summary emails returned to a user for single or batch sequence submissions. Unlike previous versions of JPred, the primary visualization of a JPred4 prediction result is a scrollable SVG image. The SVG is generated by Jalview 2.9 (www.jalview.org) (36) run in command-line mode as part of the JPred4 web server processing pipeline so users do not need to run Jalview on their own computers. However, the JalviewLite Java applet result page is still provided for users working with Java-enabled browsers who prefer direct access to Jalview's sophisticated functions.
Figure 2.

JPred4 results summary page (1) with the results of predictions presented in SVG (2). Links to detailed and simple reports in coloured HTML/PS/PDF formats (3). Example summary in HTML format is shown in (4) as well as the new addition of full multiple sequence alignments with and without gaps/insertions (5). On a separate linked page the user is able to run the Jalview applet (6) which allows a more sophisticated and interactive method of viewing the prediction results. Links to all the details for the prediction and an archive of the results are also available (7).
Figure 3.

(1) Illustration of a single sequence job submission secondary structure prediction results summary email with link to full result details (2). (3) Illustration of a batch submission email summary with overall and per job (4) details that give links to individual predictions and an archive with all results for all sequences submitted in the batch.
In all previous versions of JPred, the alignment returned showed the full-length query sequence without gaps necessary to accommodate insertions in sequences returned from the PSI-BLAST search. JPred4 introduces options to view the full multiple alignment including all residues in all sequences or download it for further analysis. For users who have local installations of Jalview (36), Jalview feature files are provided to allow easy annotation and analysis of the alignment and predictions.
In JPred3, a batch job with multiple query sequences would return separate emails for each query. JPred4 condenses these messages into a single email with a summary of success/failure for each sequence (Figure 3) in the batch and a compressed archive of all the predictions.
All JPred4 jobs are currently stored on the server for 5 days.
Time required to complete predictions
The median time for a JPred4 prediction to return results is 5 min calculated over a recent 50 000 consecutive predictions performed by end-users in the autumn of 2014. However, the server can accommodate jobs of up to 3-h duration. Most of the time is spent in the PSI-BLAST search phase which is avoided if the user submits a pre-existing MSA. MSA predictions typically return results within a few seconds.
In summary, the JPred server has been upgraded to provide a richer user experience and to include more accurate secondary structure and solvent accessibility predictions from the JNet 2.3.1 algorithm.
Acknowledgments
We thank Dr Tom Walsh for computational support.
FUNDING
Biotechnology and Biological Sciences Research Council [BB/G022686/1, BB/J019364/1 and BB/L020742/1]; Wellcome Trust [355-804783, WT092340 and WT083481]. Funding for open access charge: Wellcome Trust [106370/Z/14].
Conflict of interest statement. None declared.
REFERENCES
- 1.Chen L., Oughtred R., Berman H.M., Westbrook J. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004;20:2860–2862. doi: 10.1093/bioinformatics/bth300. [DOI] [PubMed] [Google Scholar]
- 2.Velazquez-Muriel J.A., Valle M., Santamaría-Pang A., Kakadiaris I.A., Carazo J.M. Flexible fitting in 3D-EM guided by the structural variability of protein superfamilies. Structure. 2006;14:1115–1126. doi: 10.1016/j.str.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Montelione G.T., Zheng D., Huang Y.J., Gunsalus K.C., Szyperski T. Protein NMR spectroscopy in structural genomics. Nat. Struct. Biol. 2000;7(Suppl.):982–985. doi: 10.1038/80768. [DOI] [PubMed] [Google Scholar]
- 4.Abola E., Kuhn P., Earnest T., Stevens R.C. Automation of X-ray crystallography. Nat. Struct. Biol. 2000;7(Suppl.):973–977. doi: 10.1038/80754. [DOI] [PubMed] [Google Scholar]
- 5.Gutmanas A., Alhroub Y., Battle G.M., Berrisford J.M., Bochet E., Conroy M.J., Dana J.M., Fernandez Montecelo M.A., Van Ginkel G., Gore S.P., et al. PDBe: protein data bank in Europe. Nucleic Acids Res. 2014;42:D285–D291. doi: 10.1093/nar/gkt1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kabsch W., Sander C. How good are predictions of protein secondary structure? FEBS Lett. 1983;155:179–182. doi: 10.1016/0014-5793(82)80597-8. [DOI] [PubMed] [Google Scholar]
- 8.Dor O., Zhou Y. Achieving 80% ten-fold cross-validated accuracy for secondary structure prediction by large-scale training. Proteins Struct. Funct. Genet. 2007;66:838–845. doi: 10.1002/prot.21298. [DOI] [PubMed] [Google Scholar]
- 9.Pollastri G., McLysaght A. Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics. 2005;21:1719–1720. doi: 10.1093/bioinformatics/bti203. [DOI] [PubMed] [Google Scholar]
- 10.Mooney C., Vullo A., Pollastri G. Protein structural motif prediction in multidimensional phi-psi space leads to improved secondary structure prediction. J. Comput. Biol. 2006;13:1489–1502. doi: 10.1089/cmb.2006.13.1489. [DOI] [PubMed] [Google Scholar]
- 11.Cole C., Barber J.D., Barton G.J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell R.B., Barton G.J. The limits of protein secondary structure prediction accuracy from multiple sequence alignment. J. Mol. Biol. 1993;234:951–957. doi: 10.1006/jmbi.1993.1649. [DOI] [PubMed] [Google Scholar]
- 13.Kelley L.A., MacCallum R.M., Sternberg M.J. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 2000;299:499–520. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- 14.McGuffin L.J., Jones D.T. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–881. doi: 10.1093/bioinformatics/btg097. [DOI] [PubMed] [Google Scholar]
- 15.Karplus K., Karchin R., Draper J., Casper J., Mandel-Gutfreund Y., Diekhans M., Hughey R. Combining local-structure, fold-recognition, and new fold methods for protein structure prediction. Proteins. 2003;53(Suppl. 6):491–496. doi: 10.1002/prot.10540. [DOI] [PubMed] [Google Scholar]
- 16.Rost B., Schneider R., Sander C. Protein fold recognition by prediction-based threading. J. Mol. Biol. 1997;270:471–480. doi: 10.1006/jmbi.1997.1101. [DOI] [PubMed] [Google Scholar]
- 17.Di Francesco V., Geetha V., Garnier J., Munson P.J. Fold recognition using predicted secondary structure sequences and hidden Markov models of protein folds. Proteins. 1997;1997(Suppl. 1):123–128. doi: 10.1002/(sici)1097-0134(1997)1+<123::aid-prot16>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Dalton J.A.R., Jackson R.M. An evaluation of automated homology modelling methods at low target template sequence similarity. Bioinformatics. 2007;23:1901–1908. doi: 10.1093/bioinformatics/btm262. [DOI] [PubMed] [Google Scholar]
- 19.Baker D., Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 20.Qian B., Raman S., Das R., Bradley P., McCoy A.J., Read R.J., Baker D. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–264. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradley P., Misura K.M.S., Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 22.Bystroff C., Shao Y. Fully automated ab initio protein structure prediction using I-SITES, HMMSTR and ROSETTA. Bioinformatics. 2002;18(Suppl. 1):S54–S61. doi: 10.1093/bioinformatics/18.suppl_1.s54. [DOI] [PubMed] [Google Scholar]
- 23.Srinivasan R., Rose G.D. Ab initio prediction of protein structure using LINUS. Proteins. 2002;47:489–495. doi: 10.1002/prot.10103. [DOI] [PubMed] [Google Scholar]
- 24.Lesk A.M., Lo Conte L., Hubbard T.J.P. Assessment of novel fold targets in CASP4: predictions of three-dimensional structures, secondary structures, and interresidue contacts. Proteins Struct. Funct. Genet. 2001;45:98–118. doi: 10.1002/prot.10056. [DOI] [PubMed] [Google Scholar]
- 25.Marks D.S., Colwell L.J., Sheridan R., Hopf T.A., Pagnani A., Zecchina R., Sander C. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hopf T.A., Colwell L.J., Sheridan R., Rost B., Sander C., Marks D.S. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan C.-W., Jones D.T. Using neural networks and evolutionary information in decoy discrimination for protein tertiary structure prediction. BMC Bioinformatics. 2008;9:94. doi: 10.1186/1471-2105-9-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchan D.W.A., Ward S.M., Lobley A.E., Nugent T.C.O., Bryson K., Jones D.T. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010;38:W563–W568. doi: 10.1093/nar/gkq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T., Hönigschmid P., Schafferhans A., Roos M., Bernhofer M., et al. PredictProtein-an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014;42:W337–W343. doi: 10.1093/nar/gku366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rost B., Liu J. The PredictProtein server. Nucleic Acids Res. 2003;31:3300–3304. doi: 10.1093/nar/gkg508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuff J.A., Barton G.J. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 32.Fox N.K., Brenner S.E., Chandonia J.-M. SCOPe: Structural Classification of Proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014;42:D304–D309. doi: 10.1093/nar/gkt1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzek B.E., Huang H., McGarvey P., Mazumder R., Wu C.H. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 35.Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]