

Abstract
RSAT (Regulatory Sequence Analysis Tools) is a modular software suite for the analysis of cis-regulatory elements in genome sequences. Its main applications are (i) motif discovery, appropriate to genome-wide data sets like ChIP-seq, (ii) transcription factor binding motif analysis (quality assessment, comparisons and clustering), (iii) comparative genomics and (iv) analysis of regulatory variations. Nine new programs have been added to the 43 described in the 2011 NAR Web Software Issue, including a tool to extract sequences from a list of coordinates (fetch-sequences from UCSC), novel programs dedicated to the analysis of regulatory variants from GWAS or population genomics (retrieve-variation-seq and variation-scan), a program to cluster motifs and visualize the similarities as trees (matrix-clustering). To deal with the drastic increase of sequenced genomes, RSAT public sites have been reorganized into taxon-specific servers. The suite is well-documented with tutorials and published protocols. The software suite is available through Web sites, SOAP/WSDL Web services, virtual machines and stand-alone programs at http://www.rsat.eu/.
INTRODUCTION
The Regulatory Sequence Analysis Tools (RSAT) is a software suite integrating a wide variety of programs to analyse cis-regulatory elements in genomic sequences. Since its initial development in 1998 (1,2), RSAT has provided uninterrupted service and has broadened its applications (Figure 1), following advances in the field of regulatory genomics. The suite is organized in a modular way: programs can be accessed individually or interconnected into pipelines to perform more complex analyses. The Web interface combines 52 tools enabling to perform distinct types of analyses (Table 1): obtaining sequences, discovering motifs ab initio, scanning sequences to predict transcription factor (TF) binding sites, comparing and clustering motifs, analyzing conservation and divergence of TF binding sites, detecting inter-individual regulatory variations and building control sets based on a wide variety of probabilistic models. Altogether, the RSAT Web site includes nine novel programs (tagged with asterisks in Table 1) in addition to the 43 tools described in previous NAR Web software issues (3–5). In this article, we summarize the main functionalities and novelties of the toolbox, describe the supporting teaching and training facilities and explain its various modes of access.
Figure 1.
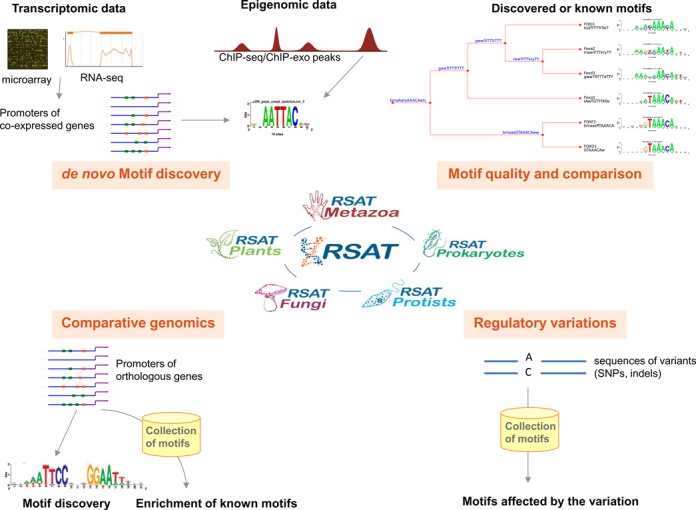
Overview of the main applications of RSAT.
Table 1. Selection of some tools available on RSAT Web servers.
| Application field | Program name | Input | Output | Description |
|---|---|---|---|---|
| Obtaining sequences (Sequence Tools) | retrieve-seq | Gene names | Sequences | Given a set of gene names, returns upstream, downstream (relative to ORF start) or unspliced ORF sequences. Segments overlapping an upstream ORF can be excluded or included. |
| * fetch-sequences (from UCSC) | Genomic coordinates | Sequences | From a set of genomic coordinates (BED file), collects the sequences from the UCSC genome browser. | |
| retrieve-ensembl-seq | Gene names | Sequences | Returns upstream, downstream, intronic, exonic, UTR, mRNA or CDS for a list of genes from Ensembl. Multi-genome queries enable automatic retrieval of sequences for gene orthologues. | |
| * retrieve-variation-seq | Identifier of variations | Sequences of the variants | Given a set of IDs for genetic variations, returns the corresponding variants and their flanking sequences. The output file can be scanned with the tool ‘variation-scan’. | |
| Motif discovery | oligo-analysis | Sequences | Over/under-represented oligonucleotides + PSSM | Analyses oligonucleotide occurrences in a set of sequences and detects over/under-represented oligonucleotides, using various background models and scoring statistics. |
| dyad-analysis | Sequences | Over/under-represented dyads + PSSM | Detects over-represented dyads (spaced pairs of oligonucleotides) within a set of sequences. | |
| NGS ChIP-seq | peak-motifs | Sequences | Discovered motifs + predicted sites | Discovers motifs in ChIP-seq peak sequence sets and returns detailed information on sequence composition and discovered motifs, with correspondences in databases and predicted binding sites. |
| Pattern matching | * crer-scan | Transcription factor binding sites | Cis-regulatory enriched regions (CRER) | Given a set of cis-regulatory elements (predicted sites, annotated sites, ChIP-seq peaks), detects regions presenting a significant enrichment in CRERs. |
| matrix-scan (-quick) | Sequences + PSSMs | Matching positions in input sequences | Scans sequences with one or several PSSMs to identify instances of the corresponding motifs (putative sites). Supports a variety of background models (Bernoulli, Markov chains of any order). | |
| * variation-scan | Variant sequences | Regulatory variants | Scans variant sequences with PSSMs and report variations that affect the binding score, in order to predict regulatory variants. | |
| dna-pattern | Sequences + patterns | Matching positions in input sequences | String-based pattern matching program specialized for DNA sequences. Supports IUPAC code for partially specified nucleotides, regular expressions and search simultaneously multiple patterns. | |
| Motif quality and comparisons (Matrix Tools) | matrix-quality | Motif (PSSM) + sequence set(s) | Score distribution statistics + ROC curves | Evaluates the quality of a PSSM by comparing score distributions obtained with this matrix in control sequence sets. |
| compare-matrices | Two sets of PSSM | Similarity scores + matrix alignments | Compares two collections of PSSMs and returns various similarity statistics + matrix alignments. | |
| * matrix-clustering | One set of PSSM | Clusters of matrices + similarity trees | Clusters similar PSSMs and builds consensus matrices for each cluster. | |
| Comparative genomics | get-orthologs | Gene names + taxon | List of homologous genes with percentage of identity, alignment length and e-value | Given a list of genes from a query organism and a reference taxon, returns the orthologues of the query gene(s) in all the organisms belonging to the reference taxon. |
| footprint-discovery | Sequences | Conserved dyads + PSSM | Detects phylogenetic footprints by applying ‘dyad-analysis’ in promoters of a set of orthologous genes. | |
| * footprint-scan | Sequences + PSSM | Conserved motifs + binding sites | Scans promoters of orthologous genes with one or several PSSMs to detect enriched motifs and predict phylogenetically conserved target genes. | |
| Building control sets | random-seq | Sequence specifications | Sequences | This program generates random sequences. Different probabilistic models are proposed (equiprobable nucleotides, specific alphabet utilization, Markov chains). |
| random-genes | Name of an organism | Genes | Selects a random set of genes in a given genome. | |
| random-genome-fragments | Name of an organism | Randomly selected genome fragments | Selects a set of fragments with random positions in a given genome supported in either RSAT or Ensembl and returns their coordinates and/or sequences. | |
| permute-matrix | One set of PSSM | Randomized PSSMs | Randomizes a set of input matrices by permuting their columns. The resulting motifs have the same nucleotide composition and information content as the original ones. |
This table only displays the most central tools available on the Web interface. See the RSAT Web site for an exhaustive list of available tools. The new tools since the 2011 Web software issue are marked with an asterisk (*).
RSAT FUNCTIONALITIES
De novo motif discovery in genome-wide data sets
RSAT core programs focus on finding putative regulatory signals by detecting exceptional motifs in a set of sequences. These sequences can correspond, for example, to regulatory regions of co-expressed genes obtained from transcriptome profiling (e.g. microarrays, RNA-seq) or regions revealed by epigenomic experiments (e.g. ChIP-seq, ChIP-exo, DNaseI, ATAC-seq) to be likely bound by a given TF or associated with open chromatin.
RSAT provides tools to retrieve promoter sequences (retrieve-seq, retrieve-ensembl-seq (6); Table 1) from a list of genes. For genome-wide epigenomic data sets, a new program extracts the sequences corresponding to a list of genomic coordinates specified in BED format (fetch-sequences from UCSC). The UCSC database is used for this task, as its programmatic access for sequences via DAS is very efficient, although it does not support repeat-masked sequences. In the future, we will add support for the new programmatic access to Ensembl via REST, which does support repeat-masked sequences.
Sequences are then used to perform ab initio motif discovery, based on a variety of criteria: over-represented oligonucleotides (oligo-analysis (1)) or spaced pairs (dyad-analysis (7)), positionally biased oligonucleotides (position-analysis (8), local-word-analysis) and differential motif representation between two data sets (oligo-diff). To facilitate analysis of genome-wide data sets, we provide a predefined pipeline (peak-motifs, (9,10)) that performs motif discovery with multiple algorithms, compares the predicted motifs with databases and enables visualization of putative binding sites in the UCSC genome browser. The computing efficiency of ‘peak-motifs’ enables online analysis of full data sets (several tens of megabases), without size restriction, within a few minutes.
The discovered motifs are usually used in a second step to scan the original set of sequences and locate putative binding sites (‘dna-pattern’, ‘matrix-scan’ (11)). TF binding sites often form clusters, potentially corresponding to enhancers. Identifying such cis-regulatory modules is achieved in RSAT by predicting cis-regulatory enriched regions (CRERs) (11). Initially embedded within ‘matrix-scan’, detection of CRERs has been re-designed as an independent program (crer-scan) to increase its computing efficiency and expand its scope. Initially limited to binding sites predicted with RSAT ‘matrix-scan’, it now takes as input any set of feature coordinates (e.g. annotated sites, ChIP-seq peaks) and detects windows significantly enriched in these features. This quicker version now enables the scanning of genome-wide data sets.
Since its early development, RSAT comprises several tools to build negative control sets, which can be used to assess the reliability of results obtained from predictive programs (random-seq, random-genes). For genome-wide analyses such as ChIP-seq, random data sets can be prepared by selecting sequences at random positions from a given genome (random-genome-fragments), or random controls can be performed by scanning the original sequences with permuted motifs (permute-matrix).
Comparing and clustering motifs
RSAT proposes an extended support for detailed analysis of motifs represented as position-specific scoring matrices (PSSMs). First, it comprises a program to assess the quality of a PSSM on user-defined sequence data sets by comparing theoretical and empirical score distributions (matrix-quality (12)). This program has also proven its usefulness to measure the enrichment of genomic regions (e.g. ChIP-seq peak sets) for one or several TF binding motifs. Second, there is an increasing demand for comparing motifs of various sources: discovered motifs versus collections of annotated matrices such as JASPAR (13), motifs discovered by different algorithms, or in different biological data sets. We have thus increased the computing efficiency of our motif comparison program (compare-matrices). Third, comparing multiple motifs and identifying clusters of similarities amongst them is very useful to regroup redundant matrices returned by several motif discovery algorithms, or to study the relationships between motifs bound by families of phylogenetically related TFs. To this purpose, we have implemented a new tool, ‘matrix-clustering’, which performs hierarchical clustering on a set of input motifs, draws trees to highlight the similarities between them (computed with ‘compare-matrices’), computes consensus motifs (matrices, IUPAC and logos) at each branch of the trees and generates a dynamic report enabling users to customize the graphical representations of motif similarities (Castro-Mondragon, J.A. et al., in preparation).
Detecting regulatory variations
Population genomics has given rise to large amounts of genetic variation data in human populations, in some model organisms and in several plants. Furthermore, for Human, Genome-Wide Association Studies (GWAS), which aim at discovering loci and genes associated with diseases, peculiar phenotypes or quantitative traits, have produced over 10 000 single nucleotide polymorphisms (SNPs) with reported trait associations, accessible via the NHGRI catalogue (14). GWAS results are also available for other organisms (https://easygwas.tuebingen.mpg.de/). Of note, a large fraction of reported SNPs are located outside of protein coding sequences (15,16). RSAT now provides a tool to extract genetic variants together with their flanking sequences (retrieve-variation-seq), which can then be scanned with a collection of motifs to predict the impact of the variation on TF binding (variation-scan). Impact on TF binding is computed by comparing the site scores (weight score difference and P-value fold change) obtained with a PSSM on sequences containing the different reported alleles for one SNP in sliding windows (Medina-Rivera, A. et al., in preparation).
Comparative genomics
The high number of sequenced prokaryotic genomes makes cross-species conservation (sequences or biological features) an increasingly powerful approach to detect potentially functional genomic elements in non-coding regions. RSAT supports both motif discovery and motif scanning approaches in the context of comparative genomic applications. Starting from a gene of interest, two predefined pipelines are provided to extract promoters of orthologous genes and discover over-represented motifs (footprint-discovery (17,18), limited to Prokaryotes and Fungi), or scan them with user-specified motifs to predict phylogenetically conserved target genes for known TFs (footprint-scan, limited to Prokaryotes and Fungi).
Compatibility with other programs and resources
As far as we know, there is no other software suite dedicated to cis-regulation and covering an as wide scope of functionalities as RSAT. The main alternative to RSAT is the MEME suite (19), which is limited to motif analyses. MEME and RSAT can be used in a complementary way, as RSAT encompasses the utility tool ‘convert-matrix’ supporting as input MEME-formatted results. RSAT actually includes several utility tools to ensure inter-conversion between alternative formats for different types of objects: ‘convert-matrix’, ‘convert-seq’, ‘convert-features’, ‘convert-variations’, ‘convert-background-model’, ‘convert-classes’ and ‘convert-graph’. These simple programs facilitate inter-connections between RSAT and complementary methods, extending the potential usages of the tools.
RSAT 2015 NOVELTIES
In addition to the novel programs described above, we have made particular efforts to facilitate the installation of the RSAT suite, in particular by packaging it in a virtual machine (see below). The other novelties mainly concern the management of supported genomes.
Taxon-specific public web sites
In order to cope with the exponential increase of available genomes, the new RSAT release presents a reconfigured organization of the public Web sites based on five servers dedicated to specific taxonomic groups. It is important to note that some types of analyses have become taxon-specific. For example, motif discovery approaches by phylogenetic footprints have proven powerful with Bacteria and Fungi taxa, but remain delusive with Metazoa. Similarly, the methods for detecting regulatory variants depend on the availability of primary data about genetic polymorphism, which is currently essentially available for a few metazoans (human and model organisms) and one yeast (Saccharomyces cerevisiae) genomes. To address the specific needs of user communities and better guide them to the appropriate tools, we set up taxon-dedicated servers, organized according to Ensembl Genomes divisions into Prokaryotes (regrouping Bacteria and Archaea), Fungi, Protists (a multi-clade grouping), Metazoa (merging metazoan species from ensembl.org, which mainly hosts vertebrates, and ensemblgenomes.org, which hosts non-vertebrate metazoans) and Plants. These taxon-specific servers also provide adapted collections of reference motifs, extracted from specialized databases: RegulonDB for Bacteria (20), JASPAR for Metazoa (13) and footprintDB for Plants (21). Furthermore, we are dedicating one server to teaching courses and tutorials (see below), which will provide access to all tools and support representative sets of organisms from each taxon.
Extension of supported organisms
In addition to the previously available organisms imported from the NCBI and Ensembl database, we have added support for Ensembl Genomes (22). As of January 2015, RSAT public servers support 3314 genomes (including 2941 Bacteria, 170 Archaea, 123 Fungi, 40 Metazoa, 18 Plants and 22 Protists).
LEARNING TO USE RSAT
We provide extensive material to help users becoming familiar with the RSAT suite.
Question-based guidance through the tools
Each server's home page now includes a dynamic menu guiding new users to the appropriate tool for his/her question, within a selection of the most common analyses.
Online help on the web pages
Each tool is documented by a manual and is equipped with one or several ‘Demo’ buttons to load the form with illustrative test cases. Some of the tools are also documented by online tutorials, to explain how to choose the relevant parameters and interpret the results. A tutorial given at the ECCB 2014 entitled ‘Analysis of Cis-Regulatory Motifs from High-Throughput Sequence Sets’ is also accessible to all users and constitutes a useful guide to the various access modes to RSAT.
Published protocols and tutorials
A series of protocols have been published (10,11,23–27) to cover some core applications of RSAT. These explain how to manipulate the tools and the underlying algorithms, and guide the reader to gain experience in the biological interpretation of the results.
Outreach and training
Since its initial development, the RSAT team has been committed to education, providing courses and workshop to students and scientific community around the world. Courses include introduction to basic pattern-matching and pattern-discovery approaches as well as application to biological questions (e.g. transcriptome analysis, microbial genome regulation, comparative bacterial genomics, ChIP-seq analysis), with a particular emphasis on ‘hands on’ data analysis. Courses material is available at http://teaching.rsat.eu/.
EXTENDED ACCESS MODES TO THE TOOLS
RSAT can be accessed in different ways: via the Web sites, SOAP Web services or the Unix command-line. In addition, RSAT can now be used through a virtual machine, installed either on a local server or on a computer cloud. We are currently working towards integrating RSAT within the Galaxy framework (28).
Web server
The simplest way to use the RSAT suite is via its Web sites, which provide a user-friendly interface and do not require any particular computational skills. The tools are organized in a modular way: at the end of each analysis, the result page proposes a list of buttons to send the results as input for the complementary tools. For example, tools producing sequences are automatically interconnected to all the tools taking sequences as input.
Web services
To use RSAT for repetitive tasks or to combine several tools into custom pipelines, we provide Web services implemented using the standards SOAP/WSDL (Simple Object Access Protocol/Web Services Description Language). For this programmatic access, users can write clients in any SOAP-supported language (e.g. Perl, Python, Java).
Virtual machine
For users wishing a local version of RSAT, the easiest option is to download the ready-to-use Virtual Machine (RSAT Download page: http://teaching.rsat.eu/download-request_form.cgi). The advantages of this solution are: (i) to run RSAT on any operating system supporting VirtualBox (including Windows); (ii) to avoid installing dependencies (libraries for the system, Perl, Python, etc.); and (iii) security, by ensuring an isolation from the host system and data space. The main drawback of this solution is the requirement of sufficient computing resources: at least 2 Gb of memory allocated to the virtual machine and 6 Gb of storage for the guest operating system (Linux Ubuntu 14.04), in addition to the RSAT package and genomes. The RSAT Virtual Machine can be installed on a cloud, as done for French users via the Cloud of the Institut Français de Bioinformatique (http://cloud.france-bioinformatique.fr).
Installing RSAT in user's operating system
The whole software suite can also be downloaded (RSAT Download page: http://teaching.rsat.eu/download-request_form.cgi) and installed on Unix-type operating systems (e.g. Linux, Mac OSX). The local installation enables to directly call each program on the command-line interpreter. This presents several advantages: (i) access more tools than presented on the Web sites; (ii) install custom collection of genomes; (iii) automate analyses by integrating the tools in custom scripts; and (iv) parallelize analyses on multi-processor configurations. The downloaded tools also enable to set up a custom Web server, to support the needs of local communities. The drawbacks of the local installation are (i) the requirement to install several programs and libraries whilst ensuring their compatibility with other local resources, and (ii) the need for substantial disk space to store the genomes of interest.
CONCLUSIONS
RSAT is possibly the most comprehensive academic suite of programs for the analysis of cis-regulatory sequences. In addition to the core motif discovery programs, which are scalable to genome-wide analyses, RSAT has been expanded to diversify its applications, including comparison and clustering of motifs, regulatory variants analyses and comparative genomics. A key strength is its interoperability with other databases (supports for many motif collections) and web tools (thanks to inter-conversions between file formats). Contrary to many programs that are dedicated to few model organisms, RSAT offers access to thousands of genomes from all kingdoms, facilitated by a new taxon-specific organization of the public servers. Its various modes of access and comprehensive documentation suit the needs of various types of users, from experimental biologists wishing to analyse their data sets without programming skills, to bioinformaticians wishing to integrate RSAT within their automated analysis workflows.
AVAILABILITY
All public RSAT servers are accessible from the RSAT portal at http://www.rsat.eu/. RSAT Web servers can be freely accessed by all users without login requirement.
Acknowledgments
We are particularly thankful to the colleagues who help us in installing and maintaining RSAT servers: Victor del Moral Chavez, Romualdo Zayas-Lagunas, Alfredo José Hernández Alvarez (Centro de Ciencias Genomicas, Cuernavaca, Mexico) and Erik Bongcam-Rudloff (BMC, Uppsala, Sweden). We especially acknowledge Julio Collado-Vides, who initiated the project in 1997 and supported it during the last 17 years. We thank Mauricio Guzman for designing all logos for RSAT and Benjamin Bardiaux for valuable technical ideas. We acknowledge Lionel Spinelli for his valuable advice about software design and his active participation in discussions about RSAT organization.
FUNDING
The EU-Funded COST Action [BM1006 ‘SEQAHEAD—Next Generation Sequencing Data Analysis Network’]; FP7 MICROME Collaborative Project [222886-2]; Programa Euroinvestigación/Plant KBBE 2008 [EUI2008-03612 to B.C.M.]; the GIS IBiSA and France Génomique National Infrastructure, ‘Investissements d'Avenir’, the French Agence Nationale pour la Recherche [ANR-10-INBS-09]; Institut Français de Bioinformatique National Infrastructure, ‘Investissements d'Avenir’, the French Agence Nationale pour la Recherche [ANR-11-INBS-0013]; iBone [ANR-13-EPIG-0001-04]; EchiNodal [ANR-14-CE11-0006-02]; CONACyT-Mexico and Contrat Doctoral d'Aix-Marseille Université attribué sur Concours EDSVS [to J.C.M.]; the Consejo Nacional de Ciencia y Tecnología (CONACYT) [to A.M.R.]; CIHR Training Grant in Genetic Epidemiology and Statistical Genetics [GET-101831 to A.M.R.]. Funding for open access charge: Institut National de la Santé et de la Recherche Médicale (Inserm).
Conflict of interest statement. None declared.
REFERENCES
- 1.van Helden J., André B., Collado-Vides J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J. Mol. Biol. 1998;281:827–842. doi: 10.1006/jmbi.1998.1947. [DOI] [PubMed] [Google Scholar]
- 2.van Helden J., Collado-Vides J., André B., Andr B. A web site for the computational analysis of yeast regulatory sequences. Yeast. 2000;16:177–187. doi: 10.1002/(SICI)1097-0061(20000130)16:2<177::AID-YEA516>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 3.van Helden J. Regulatory sequence analysis tools. Nucleic Acids Res. 2003;31:3593–3596. doi: 10.1093/nar/gkg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas-Chollier M., Sand O., Turatsinze J.-V., Janky R., Defrance M., Vervisch E., Brohée S., van Helden J. RSAT: regulatory sequence analysis tools. Nucleic Acids Res. 2008;36:W119–W127. doi: 10.1093/nar/gkn304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas-Chollier M., Defrance M., Medina-Rivera A., Sand O., Herrmann C., Thieffry D., van Helden J. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res. 2011;39:W86–W91. doi: 10.1093/nar/gkr377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sand O., Thomas-Chollier M., van Helden J. Retrieve-ensembl-seq: user-friendly and large-scale retrieval of single or multi-genome sequences from Ensembl. Bioinformatics. 2009;25:2739–2740. doi: 10.1093/bioinformatics/btp519. [DOI] [PubMed] [Google Scholar]
- 7.van Helden J., Rios A.F., Collado-Vides J. Discovering regulatory elements in non-coding sequences by analysis of spaced dyads. Nucleic Acids Res. 2000;28:1808–1818. doi: 10.1093/nar/28.8.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Helden J., del Olmo M., Pérez-Ortín J.E. Statistical analysis of yeast genomic downstream sequences reveals putative polyadenylation signals. Nucleic Acids Res. 2000;28:1000–1010. doi: 10.1093/nar/28.4.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas-Chollier M., Herrmann C., Defrance M., Sand O., Thieffry D., van Helden J. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res. 2012;40:e31. doi: 10.1093/nar/gkr1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas-Chollier M., Darbo E., Herrmann C., Defrance M., Thieffry D., van Helden J. A complete workflow for the analysis of full-size ChIP-seq (and similar) data sets using peak-motifs. Nat. Protoc. 2012;7:1551–1568. doi: 10.1038/nprot.2012.088. [DOI] [PubMed] [Google Scholar]
- 11.Turatsinze J.-V., Thomas-Chollier M., Defrance M., van Helden J. Using RSAT to scan genome sequences for transcription factor binding sites and cis-regulatory modules. Nat. Protoc. 2008;3:1578–1588. doi: 10.1038/nprot.2008.97. [DOI] [PubMed] [Google Scholar]
- 12.Medina-Rivera A., Abreu-Goodger C., Thomas-Chollier M., Salgado H., Collado-Vides J., van Helden J. Theoretical and empirical quality assessment of transcription factor-binding motifs. Nucleic Acids Res. 2010;39:808–824. doi: 10.1093/nar/gkq710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.-Y., Chou A., Ienasescu H., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2013;42:D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X., Zhu C., Yeh C.-T., Wu W., Takacs E.M., Petsch K.A., Tian F., Bai G., Buckler E.S., Muehlbauer G.J., et al. Genic and nongenic contributions to natural variation of quantitative traits in maize. Genome Res. 2012;22:2436–2444. doi: 10.1101/gr.140277.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janky R., van Helden J. Evaluation of phylogenetic footprint discovery for predicting bacterial cis-regulatory elements and revealing their evolution. BMC Bioinformatics. 2008;9:37. doi: 10.1186/1471-2105-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brohée S., Janky R., Abdel-Sater F., Vanderstocken G., André B., van Helden J. Unraveling networks of co-regulated genes on the sole basis of genome sequences. Nucleic Acids Res. 2011;39:6340–6358. doi: 10.1093/nar/gkr264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salgado H., Peralta-Gil M., Gama-Castro S., Santos-Zavaleta A., Muñiz-Rascado L., García-Sotelo J.S., Weiss V., Solano-Lira H., Martínez-Flores I., Medina-Rivera A., et al. RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res. 2013;41:D203–D213. doi: 10.1093/nar/gks1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sebastian A., Contreras-Moreira B. footprintDB: a database of transcription factors with annotated cis elements and binding interfaces. Bioinformatics. 2014;30:258–265. doi: 10.1093/bioinformatics/btt663. [DOI] [PubMed] [Google Scholar]
- 22.Kersey P.J., Allen J.E., Christensen M., Davis P., Falin L.J., Grabmueller C., Hughes D.S.T., Humphrey J., Kerhornou A., Khobova J., et al. Ensembl Genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res. 2014;42:D546–D552. doi: 10.1093/nar/gkt979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janky R., van Helden J. Discovery of conserved motifs in promoters of orthologous genes in prokaryotes. Methods Mol. Biol. 2007;395:293–308. doi: 10.1007/978-1-59745-514-5_18. [DOI] [PubMed] [Google Scholar]
- 24.Sand O., van Helden J. Discovery of motifs in promoters of coregulated genes. Methods Mol. Biol. 2007;395:329–348. doi: 10.1007/978-1-59745-514-5_21. [DOI] [PubMed] [Google Scholar]
- 25.Brohée S., Faust K., Lima-Mendez G., Vanderstocken G., van Helden J. Network Analysis Tools: from biological networks to clusters and pathways. Nat. Protoc. 2008;3:1616–1629. doi: 10.1038/nprot.2008.100. [DOI] [PubMed] [Google Scholar]
- 26.Defrance M., Janky R., Sand O., van Helden J. Using RSAT oligo-analysis and dyad-analysis tools to discover regulatory signals in nucleic sequences. Nat. Protoc. 2008;3:1589–1603. doi: 10.1038/nprot.2008.98. [DOI] [PubMed] [Google Scholar]
- 27.Sand O., Thomas-Chollier M., Vervisch E., van Helden J. Analyzing multiple data sets by interconnecting RSAT programs via SOAP Web services: an example with ChIP-chip data. Nat. Protoc. 2008;3:1604–1615. doi: 10.1038/nprot.2008.99. [DOI] [PubMed] [Google Scholar]
- 28.Goecks J., Nekrutenko A., Taylor J., Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]