


Abstract
Macromolecular interactions play a crucial role in biological systems. Simulation of diffusional association (SDA) is a software for carrying out Brownian dynamics simulations that can be used to study the interactions between two or more biological macromolecules. webSDA allows users to run Brownian dynamics simulations with SDA to study bimolecular association and encounter complex formation, to compute association rate constants, and to investigate macromolecular crowding using atomically detailed macromolecular structures. webSDA facilitates and automates the use of the SDA software, and offers user-friendly visualization of results. webSDA currently has three modules: ‘SDA docking’ to generate structures of the diffusional encounter complexes of two macromolecules, ‘SDA association’ to calculate bimolecular diffusional association rate constants, and ‘SDA multiple molecules’ to simulate the diffusive motion of hundreds of macromolecules. webSDA is freely available to all users and there is no login requirement. webSDA is available at http://mcm.h-its.org/webSDA/.
INTRODUCTION
Macromolecular interactions, such as protein–protein interactions and protein–DNA interactions, are key to the function of biological systems. Studying macromolecular diffusional association computationally can give insights into biological interactions. Simulation of diffusional association (SDA) is a Brownian dynamics simulation software which can be used to perform macromolecular docking, to compute bimolecular association rate constants, and to simulate molecular diffusion for systems of many macromolecules (1–3). Intermolecular forces are computed from atomic-detail molecular structures. SDA has been used to generate diffusional encounter complexes for a wide range of macromolecular complexes, including those of barnase and barstar (2,4), cytochrome P450 enzymes and their redox protein partners (5–7), methionine aminopeptidase and the ribosome (8), and the linker histone H5 and the nucleosome (9). Moreover, SDA has been used to calculate association rate constants (1,2,10,11) and simulate the diffusive motion of hundreds of macromolecules in solution (3,12–15). All these functions of SDA have been extensively validated against experimental data. Although SDA is made available for standalone use, it cannot be used easily by non-experts because of its multitude of different parameters and options, many of which are interdependent. Setting these parameters correctly and understanding their function demands a good understanding of Brownian dynamics simulations and the structure of the SDA software.
webSDA is a web server which aims to eliminate these obstacles and make SDA's functionalities available to a broader audience. webSDA supports the generation of diffusional encounter complexes (‘SDA docking’), the calculation of biomolecular diffusional association rate constants (‘SDA association’) and the simulation of the diffusive motion of several hundred macromolecules (‘SDA multiple molecules’). The flexibility of the solutes can be accounted for by providing several different conformations for one of the solutes (‘SDA docking’ and ‘SDA association’) or each of the solutes (‘SDA multiple molecules’). It allows users to set up and run SDA simulations. webSDA automates the preparation of input files by generating input parameters automatically. If one parameter is used in multiple input files, it only needs to be defined once. Input files generated by webSDA can also be downloaded for use with the standalone version of SDA.
webSDA WORKFLOW
The workflow of webSDA is shown in Figure 1.
Figure 1.
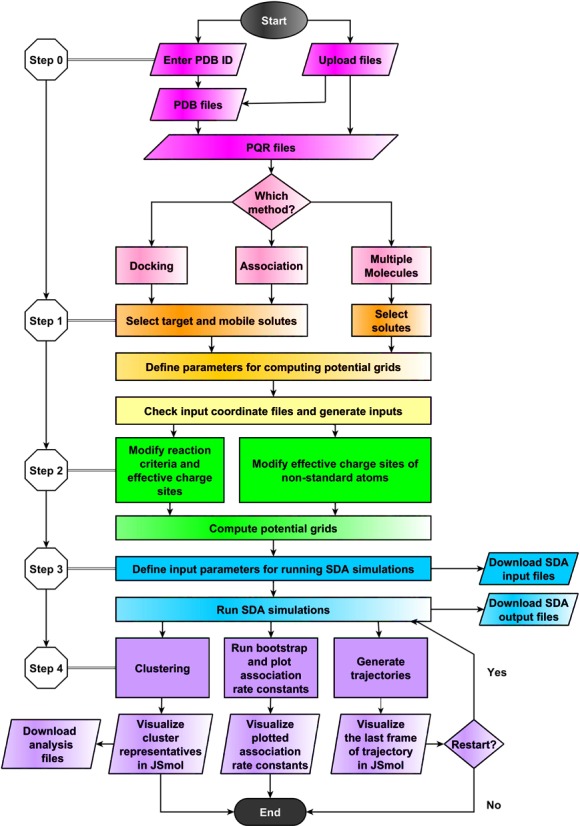
Workflow of webSDA.
Step 0
To begin a simulation, users may enter the PDB identifiers of the macromolecules that they wish to simulate or upload coordinate files for the macromolecules in PDB or PQR format. PQR format files contain the atomic coordinates, formatted as in PDB files from the Protein Data Bank (www.rcsb.org/pdb), with partial atomic charges (Q) and atomic radii (R) instead of B-factors and occupancies. If PDB files are used, PQR files will be generated using the PDB2PQR software, version 2.0.0 (16), with the AMBER force field and default parameters. Users can also generate their own PQR files using the PDB2PQR web server (16). After uploading coordinate files, users select one of the SDA methods: ‘SDA docking’, ‘SDA association’ or ‘SDA multiple molecules’. Users then follow a sequence of steps to set up and run an SDA job, guided by hints on the webSDA website. As an example, the following steps show how to run an ‘SDA docking’ simulation.
Step 1
Select target and mobile solutes: users need to choose a target solute and a mobile solute for the ‘SDA docking’ job.
Step 2
Modify reaction criteria and effective charge sites: reaction criteria are distances between defined points in the solute molecules that are required to be satisfied for an encounter complex to be recorded in an SDA simulation. The default reaction criterion for ‘SDA docking’ is predefined for the centers of geometry of the solutes and requires that they approach within a distance equal to the sum of the maximum extensions of the two solutes plus 12 Å. The reaction criteria can be changed in the web interface and can be used to impose additional distance constraints which can be derived, e.g. from bioinformatics analysis or experimental data, such as FRET and mutagenesis. Effective charges are used in combination with electrostatic potential grids to approximate the electrostatic interactions between molecules (17). Users can modify the effective charge sites for non-standard atoms, such as found in low-molecular weight compounds, in the web interface.
Step 3
Define input parameters for running SDA simulations: the automatically generated SDA parameters are given to users. Users can modify the SDA input parameters and run SDA simulations on the server. webSDA checks the input parameters that users have changed and helps users to set correct parameters. They may upload their own files (SDA input file, reaction criteria file and effective charge sites file) to run their SDA simulations on the web server. The web server can be used to run short jobs. The SDA input file can also be generated and downloaded together with the other files to run with the standalone version of the SDA software, which is also freely available.
Step 4
Clustering: for ‘SDA docking’, the recorded diffusional encounter complexes are clustered and the cluster representatives can be visualized with JSmol on the web server or downloaded as described below.
OUTPUT
Besides the option to download output files, webSDA offers a convenient visualization of the results. An example is given in Figure 2. For the results of ‘SDA docking’, JSmol is used for visualization of the cluster representatives of the diffusional encounter complexes. This provides users with an easy way to view encounter complexes and decide which complexes to download for further analyses. For ‘SDA association’, the calculated association rate constants are plotted using the R language, enabling users to visualize calculated association rate constants immediately. For ‘SDA multiple molecules’, the JSmol plug-in is employed to enable users to visualize the final snapshot of their simulations.
Figure 2.
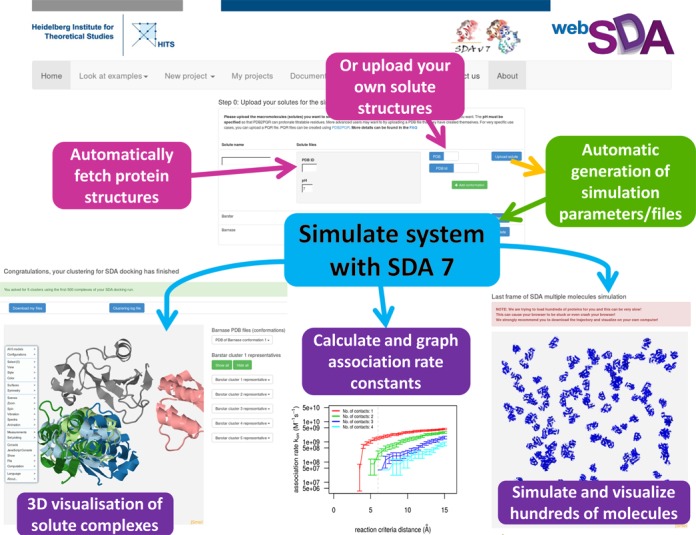
Screenshots of webSDA showing key features of webSDA.
webSDA assigns each user a universally unique identifier (UUID). This enables users to revisit all projects that they have run in the ‘My projects’ page. There is a link associated with this page and users can send this link to colleagues to share projects.
EXAMPLES OF webSDA USAGE
webSDA gives results consistent with the standalone version of SDA, which has been extensively validated against experimental data (1,8,12). webSDA provides example cases for each method. In Figure 2, we illustrate the performance of webSDA for the simulation of the association of barnase and barstar (PDB ID: 1BRS) and the simulation of many barnase molecules. With the ‘SDA docking’ method, cluster representatives of the docked barnase-barstar complexes in good agreement with the crystal structure of the complex and results obtained by Motiejunas et al. (4) are obtained.
It should be kept in mind that ‘SDA docking’ uses Brownian dynamics simulations to generate diffusional encounter complexes rather than fully docked complexes. The encounter complexes can be refined by molecular dynamics simulation to generate fully bound complexes, as shown in (4), but currently webSDA does not support running molecular dynamics simulation.
The ‘SDA association’ function is another important function of webSDA. For the barnase and barstar complex at an ionic strength of 50 mM and pH 7, the association rate constant computed with ‘SDA association’ is 4.1 × 108 M−1s−1 (averaged from three webSDA runs: 4.0 × 108, 4.0 × 108 and 4.3 × 108 M−1s−1), which is comparable with experimental data (3 × 108 M−1s−1) (18) (Figure 2) and the simulation results reported by Gabdoulline and Wade (1).
The ‘SDA multiple molecules’ function of webSDA can be used to perform Brownian dynamics simulations of many molecules. This function of SDA has been used to study the behaviour of proteins in crowded environments and is shown to be able to reproduce experimental data (3,12–15). The long-time self-diffusion coefficients, second virial coefficients and structure factors, calculated by ‘SDA multiple molecules’, agree with experimental values.
There are several web servers (19–24) providing similar functionality to ‘SDA docking’ but they are often limited to proteins with standard amino acid residues. ‘SDA docking’ supports protein, DNA and RNA, and small molecules, such as a cofactor, can be treated as part of a macromolecular structure. For the calculation of association rate constants, the TransComp server (http://pipe.sc.fsu.edu/transcomp/) (25) computes association rate constants for protein–protein and protein–RNA complexes. ‘SDA multiple molecules’ is, to the best of our knowledge, the first web server for performing simulations of hundreds to thousands of macromolecules, which can be used to study concentrated protein solutions, and to investigate macromolecular crowding, or other phenomena involving large numbers of macromolecules.
TECHNICAL OVERVIEW
The webSDA interface uses Javascript and HTML5 to control user interface (UI) elements. The server uses the Play framework version 2 and Apache commons.exec to manage user sessions and server resources. PDB2PQR version 2.0.0 is used to convert PDB files to PQR files (16,26). BioJava is used to process PDB ID and download PDB files. Python scripts (Biopython) are used to check PQR files and generate input parameters. SDA 7 (http://mcm.h-its.org/sda7/) is used to process PQR files, to generate grids (along with the Adaptive Poisson–Boltzmann Software (27) to compute electrostatic potential grids), and to perform simulations.
Currently, the maximum run-time of a job on the webserver is 24 h and the output files for SDA runs that execute for this time are written and provided to users. A queuing system using the Terascale Open-source Resource and QUEue Manager (TORQUE) is used to manage the submission of webSDA jobs to a backend compute cluster.
DISCUSSION AND OUTLOOK
webSDA is a web server to perform Brownian dynamics simulations of biomacromolecules. ‘SDA docking’ allows users to generate diffusional encounter complexes of two solutes (proteins, DNA or RNA). ‘SDA association’ can be used to compute the bimolecular association rate constant for a specified complex and can be combined with ‘SDA docking’ when the structure of the complex is unknown. ‘SDA multiple molecules’ is helpful for studying the diffusive behaviour of hundreds of molecules.
webSDA automates the widely used SDA software, generates input parameters and files automatically, and offers user-friendly visualization of output results. webSDA is useful for beginners learning how to perform Brownian dynamics simulations as well as for experts preparing input files for running simulations. Simulation times and file sizes are limited on the webSDA server so webSDA allows the generated input files to be downloaded for carrying out production runs with SDA on local or high performance computing resources. We believe that webSDA will be an easy-to-use tool for users who are interested in using Brownian dynamics simulations to study biological systems. Currently, webSDA does not support all the functions of SDA. However, webSDA has a modular structure that will facilitate the implementation of further methods and features in the future.
Acknowledgments
X.Y. acknowledges the support of the Hartmut Hoffmann-Berling International Graduate School (HBIGS) of Molecular and Cellular Biology, Heidelberg University. We thank the members of the Molecular and Cellular Modeling group at HITS for testing of webSDA. This paper reflects only the authors’ views and neither the BMBF the IMI nor the European Commission is liable for any use that may be made of the information contained herein.
FUNDING
Klaus Tschira Foundation and the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) Virtual Liver Network [0315749]; EU/EFPIA Innovative Medicines Initiative (IMI) Joint Undertaking, K4DD [115366]; EU FEP Flagship Programme Human Brain Project [604102]. Funding for open access charge: Heidelberg Institute for Theoretical Studies.
Conflict of interest statement. None declared.
REFERENCES
- 1.Gabdoulline R.R., Wade R.C. Simulation of the diffusional association of barnase and barstar. Biophys. J. 1997;72:1917–1929. doi: 10.1016/S0006-3495(97)78838-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabdoulline R.R., Wade R.C. Brownian dynamics simulation of protein-protein diffusional encounter. Methods. 1998;14:329–341. doi: 10.1006/meth.1998.0588. [DOI] [PubMed] [Google Scholar]
- 3.Mereghetti P., Gabdoulline R.R., Wade R.C. Brownian dynamics simulation of protein solutions: structural and dynamical properties. Biophys. J. 2010;99:3782–3791. doi: 10.1016/j.bpj.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motiejunas D., Gabdoulline R., Wang T., Feldman-Salit A., Johann T., Winn P.J., Wade R.C. Protein-protein docking by simulating the process of association subject to biochemical constraints. Proteins. 2008;71:1955–1969. doi: 10.1002/prot.21867. [DOI] [PubMed] [Google Scholar]
- 5.Wade R.C., Motiejunas D., Schleinkofer K., Sudarko Winn, P.J., Banerjee A., Kariakin A., Jung C. Multiple molecular recognition mechanisms. Cytochrome P450—a case study. Biochim. Biophys. Acta. 2005;1754:239–244. doi: 10.1016/j.bbapap.2005.07.044. [DOI] [PubMed] [Google Scholar]
- 6.Karyakin A., Motiejunas D., Wade R.C., Jung C. FTIR studies of the redox partner interaction in cytochrome P450: the Pdx-P450cam couple. Biochim. Biophys. Acta. 2007;1770:420–431. doi: 10.1016/j.bbagen.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Madrona Y., Hollingsworth S.A., Tripathi S., Fields J.B., Rwigema J.C.N., Tobias D.J., Poulos T.L. Crystal structure of cindoxin, the P450cin redox partner. Biochemistry. 2014;53:1435–1446. doi: 10.1021/bi500010m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandikci A., Gloge F., Martinez M., Mayer M.P., Wade R., Bukau B., Kramer G. Dynamic enzyme docking to the ribosome coordinates N-terminal processing with polypeptide folding. Nat. Struct. Mol. Biol. 2013;20:843–850. doi: 10.1038/nsmb.2615. [DOI] [PubMed] [Google Scholar]
- 9.Pachov G.V, Gabdoulline R.R., Wade R.C. On the structure and dynamics of the complex of the nucleosome and the linker histone. Nucleic Acids Res. 2011;39:5255–5263. doi: 10.1093/nar/gkr101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabdoulline R.R., Wade R.C. Protein-protein association: investigation of factors influencing association rates by brownian dynamics simulations. J. Mol. Biol. 2001;306:1139–1155. doi: 10.1006/jmbi.2000.4404. [DOI] [PubMed] [Google Scholar]
- 11.ElSawy K.M., Caves L.S.D., Twarock R. The impact of viral RNA on the association rates of capsid protein assembly: Bacteriophage MS2 as a case study. J. Mol. Biol. 2010;400:935–947. doi: 10.1016/j.jmb.2010.05.037. [DOI] [PubMed] [Google Scholar]
- 12.Mereghetti P., Wade R.C. Atomic detail Brownian dynamics simulations of concentrated protein solutions with a mean field treatment of hydrodynamic interactions. J. Phys. Chem. B. 2012;116:8523–8533. doi: 10.1021/jp212532h. [DOI] [PubMed] [Google Scholar]
- 13.Balbo J., Mereghetti P., Herten D.-P., Wade R.C. The shape of protein crowders is a major determinant of protein diffusion. Biophys. J. 2013;104:1576–1584. doi: 10.1016/j.bpj.2013.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mereghetti P., Wade R.C. Brownian dynamics simulation of protein diffusion in crowded environments. AIP Conf. Proc. 2013;1518:511–516. [Google Scholar]
- 15.Mereghetti P., Kokh D., McCammon J.A., Wade R.C. Diffusion and association processes in biological systems: theory, computation and experiment. BMC Biophys. 2011;4:2. doi: 10.1186/2046-1682-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabdoulline R.R., Wade R.C. Effective charges for macromolecules in solvent. J. Phys. Chem. 1996;100:3868–3878. [Google Scholar]
- 18.Schreiber G., Fersht A.R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 19.De Vries S.J., van Dijk M., Bonvin A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 20.Comeau S.R., Gatchell D.W., Vajda S., Camacho C.J. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:96–99. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macindoe G., Mavridis L., Venkatraman V., Devignes M.-D., Ritchie D.W. HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010;38:W445–W449. doi: 10.1093/nar/gkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierce B.G., Wiehe K., Hwang H., Kim B.-H., Vreven T., Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu097. doi:10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torchala M., Moal I.H., Chaleil R.A.G., Fernandez-Recio J., Bates P.A. SwarmDock: a server for flexible protein-protein docking. Bioinformatics. 2013;29:807–809. doi: 10.1093/bioinformatics/btt038. [DOI] [PubMed] [Google Scholar]
- 24.Jiménez-García B., Pons C., Fernández-Recio J. pyDockWEB: a web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics. 2013;29:1698–1699. doi: 10.1093/bioinformatics/btt262. [DOI] [PubMed] [Google Scholar]
- 25.Qin S., Pang X., Zhou H.-X. Automated prediction of protein association rate constants. Structure. 2011;19:1744–1751. doi: 10.1016/j.str.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G., Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:522–525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]