


Abstract
IMP (Integrative Multi-species Prediction), originally released in 2012, is an interactive web server that enables molecular biologists to interpret experimental results and to generate hypotheses in the context of a large cross-organism compendium of functional predictions and networks. The system provides biologists with a framework to analyze their candidate gene sets in the context of functional networks, expanding or refining their sets using functional relationships predicted from integrated high-throughput data. IMP 2.0 integrates updated prior knowledge and data collections from the last three years in the seven supported organisms (Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Danio rerio, Caenorhabditis elegans, and Saccharomyces cerevisiae) and extends function prediction coverage to include human disease. IMP identifies homologs with conserved functional roles for disease knowledge transfer, allowing biologists to analyze disease contexts and predictions across all organisms. Additionally, IMP 2.0 implements a new flexible platform for experts to generate custom hypotheses about biological processes or diseases, making sophisticated data-driven methods easily accessible to researchers. IMP does not require any registration or installation and is freely available for use at http://imp.princeton.edu.
INTRODUCTION
Biologists using modern experimental methods are generating massive amounts of genome-scale data. However, there continues to be a substantial gap between the avalanche of genomic data, which are abundant but not reliable, and our limited biological knowledge, which can only be discovered through careful, small-scale techniques. This disparity has been exacerbated with the development and popularity of next-generation technologies, such as RNA-seq, which enable genome-wide measurements at unprecedented resolution and cost (1). A paucity of biological knowledge (i.e. experimentally validated gene function) limits the coverage and accuracy of computational methods that require prior knowledge to learn novel biology, even when large-scale genomic data are available (2). Thus, these methods are limited to performing well on processes and pathways that are already well characterized in an organism. IMP (Integrated Multi-species Prediction) was originally developed to address the growing need to interpret and analyze results from genome-wide studies and generate hypotheses for experimental follow-up in the context of integrated functional gene networks, even when prior knowledge is limited in an organism or for a specific biological context (3).
IMP is an exploratory tool that provides a high-quality, interactive interface for functional prediction and interrogation. Researchers can incorporate IMP into their analysis workflow in several ways. For example, biologists can overlay their genes from a high-throughput experiment onto IMP's functional gene networks, expanding or contracting the network and identifying enriched, unifying functional themes. Alternatively, researchers can generate specific functional hypotheses by querying IMP's collection of gene-pathway predictions, identifying candidate genes for a biological context of interest. In all of these analyses, IMP systematically applies a previously developed network-based method that identifies functionally similar homologs to transfer annotations (i.e. gene-pathway membership) between organisms. This more specific homology detection method extends beyond simple annotation transfer by sequence similarity and enables accurate gene pathway predictions, even for processes that have few or no experimental annotations in an organism (2).
There are several successful web servers that allow researchers to analyze their gene sets in the context of gene networks (4–6), however, they are either constrained by the availability of prior knowledge in an organism and biological process of interest or limited to sequence-based transfers of input data (7,8). IMP is unique in its systematic incorporation of a functional genomics-based homology transfer of prior knowledge (9) in all of its analyses, improving the accuracy and coverage of functional interrogation (2).
IMP has been continuously maintained and developed since the original publication and here we describe major updates to the server. We have extensively updated the gene networks and functional predictions across all seven organisms, adding publicly available gene expression experiments from the subsequent years, and updating the already included data sources. Additionally, we extend IMP's functional coverage to include human diseases, allowing biologists to analyze disease contexts and predictions in human and across model organisms. Human disease gene knowledge is transferred to other organisms and predictions are made using each organism's gene network. By exploring disease gene predictions across the model organisms, biologists can find candidate genes to serve as targets for follow-up in human and in potential animal models for their disease of interest.
Additionally, we have created a flexible tool that furthers the original goal of the web server: to enable biologists to analyze their experimental results in the context of massive-scale integrated data compendia. We developed a prediction platform that allows biologists to bring their larger experimental result (for example, a list of hundreds of genes identified as over-expressed in a microarray experiment) and run a sophisticated machine-learning method for classification. This tool can be used to answer many pertinent questions, for example, identifying additional candidate disease genes from a microarray experiment, or additional players in a biological process of interest. Such an analysis might otherwise be infeasible due to biologists’ limited computational resources or expertise. The software is maintained and executed on IMP's servers and only requires a list of genes from the user. Genome-wide results are available by email, if provided, or directly on the web server.
IMP SYSTEM DATA UPDATES
IMP is a flexible tool that can be used to answer diverse biological questions posed in the form of a biological context (a process or a disease), a single gene, or a set of genes of interest. These questions can be broad and exploratory, for example, determining the shared pathways among a set of genes that are co-expressed in an mRNA experiment. Alternatively, researchers can generate specific experimentally testable hypotheses, such as identifying functional partners of a gene of interest or possible pathways that the gene participates in. As an exploratory tool, IMP provides interactive visualizations of gene-gene functional relationships, gene-process predictions and cross-organism network alignments. IMP is both a collection of gene-pathway predictions that users can query for specific targeted results and a suite of user-driven tools that can be customized for broad discovery.
All of IMP's diverse analyses leverage an organism's functional gene network, which integrates thousands of genome-wide experiments from an array of public data sources (10–13) and describes whether genes participate in similar biological processes. These networks are constructed using previously described methods (2,6,14) and have been extensively updated in the subsequent years since IMP was originally released. We use a new expert-curated set of Gene Ontology (GO) terms (15) to define the gold standard for learning gene–gene functional relationships and have updated the standard to include the latest experimental annotations. IMP networks now integrate 3540 data sets, a 49% increase in the number of data sets from IMP's original release (3), and include over 70 000 experimental conditions. In addition to adding gene expression experiments from the last three years, IMP networks have been updated with the most recent releases of popular functional genomic databases. For example, BioGRID (10) has been updated to include 196 909 additional protein–protein interactions, an increase of 78% from the original networks. A complete list of data sources is available directly on the web server.
DISEASE PREDICTIONS
Biologists can query IMP with a gene set or biological context of interest to retrieve putative gene-pathway assignments. We have extended IMP's biological contexts to include human diseases, in addition to GO biological processes. Biologists can now analyze disease contexts and predictions across organisms. IMP applies the same machine-learning method for predicting genes to biological processes (2,3) as it does to diseases, which uses a functional network as input to a Support Vector Machine (SVM) to classify genes (Figure 1). We showed previously that this method is accurate and competitive among state-of-the-art methods in predicting genes to biological processes (2,3). Disease gene predictions are inferred directly in human—from disease genes curated by Online Mendelian Inheritance in Man (OMIM) (16) and using the human functional network—and in the six model organisms. The disease predictions inferred in the other organisms leverage biological knowledge from human by transferring OMIM knowledge using our previously described method to identify the appropriate homologs for gene annotation transfer (2,9). These human-transferred gene-disease annotations are then used as training data for prediction with the organisms’ functional network, and the subsequent gene predictions are specific to that organism. By applying a model organism's functional network to predict disease genes, IMP can help biologists address an important challenge in the study of human disease: identifying the best model system for a given disease and the appropriate orthologs for a disease of interest.
Figure 1.
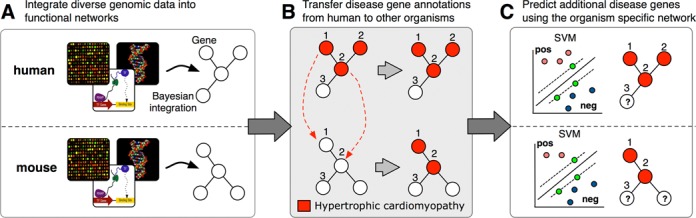
A schematic for IMP disease knowledge transfer and prediction. (A) IMP constructs a functional network for each of seven organisms by integrating heterogeneous genomic data. (B) Disease-gene annotations from human are transferred to the functionally similar homologs in other organisms. (C) Additional disease genes are predicted using the human-transferred disease genes in the organism-specific functional networks.
Using IMP, users search by Disease Ontology (DO) (17) term or by gene to retrieve gene-disease predictions. OMIM disease genes are mapped to DO, using the mapping provided by DO, to leverage the unified naming and hierarchical structure of the ontology. Figure 2 shows queries for hypertrophic cardiomyopathy (HCM) in both human (Figure 2A) and mouse (Figure 2B). Many of the top genes in the human query are known to be involved in the disease (highlighted rows), and the others are potential disease candidates. For example, the second novel gene prediction is TRIM63, which encodes an E3 ubiquitin ligase and plays a role in the atrophy of skeletal and cardiac muscle (18,19). The gene has recently been suggested (independent of IMP) as a candidate for HCM with several mutations observed in patients with the disease (20).
Figure 2.
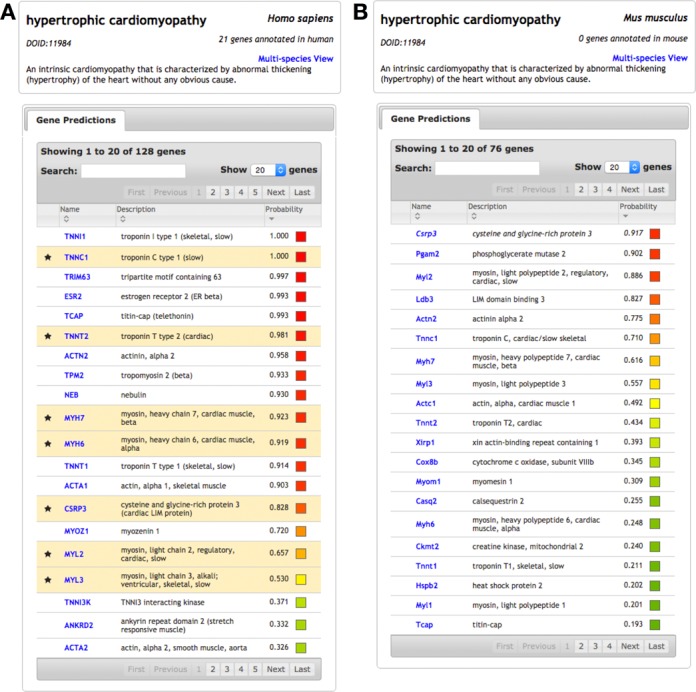
Disease result pages for ‘hypertrophic cardiomyopathy’ in IMP. (A) Querying ‘hypertrophic cardiomyopathy’ in human returns a list of genes predicted to be involved in the disease, sorted by probability. IMP applies known hypertrophic cardiomyopathy genes in human (from OMIM) to predict additional genes from the human functional network. (B) The same disease query can be performed in mouse, returning predicted mouse genes. These predictions were learned using human disease genes transferred to mouse with the mouse functional network.
Figure 2B shows the same query for HCM in mouse. These gene predictions, which leverage human disease knowledge transferred to mouse, are potentially informative as a mouse model for the disease. In fact, the most confidently predicted gene, Csrp3, was a target in the first model for dilated cardiomyopathy with hypertrophy in a genetically manipulatable organism. Csrp3-deficient mice reproduce the same morphologic and clinical features of the disease as in human (21). The Csrp3 mouse model serves as a valuable resource for understanding the pathophysiology of heart failure and for identifying potential therapies for the disease (22,23). Thus, in these example use cases, IMP independently, and in a data-driven predictive fashion, identifies a candidate human gene for HCN and a mouse gene that is already a model for understanding human HCM.
PLATFORM FOR CUSTOM PREDICTIONS
Many biological questions cannot be posed as a predefined gene set, such as a GO biological process or OMIM disease, or expressed as a small gene set (i.e. <50 genes), requiring more advanced and flexible data-mining techniques. For example, a researcher with results from a genetic screen may be interested in identifying additional candidate genes. Alternatively, a biologist may want to combine her private experimental result with public gene pathway annotations to make customized predictions. Most biologists lack the computational resources or expertise to implement and support the necessary machine learning software and data compendia for such an analysis. With IMP 2.0, we provide a flexible platform for researchers to run state-of-the-art machine learning methods and pose customized, sophisticated biological questions.
Users provide a gold standard, in the form of a set of relevant genes, or use IMP provided gene sets, which include GO biological process and DO terms. IMP uses the same previously described and validated method for predicting GO function (2,3), which applies a SVM with features constructed from the organism of interest's functional gene network for classification. The SVM classifies all genes in the genome based on its pattern of functional relationships with the provided genes of interest, up-weighting the parts of the network that are informative for membership in the gene set. This method has been previously shown to be accurate in predicting genes to biological processes and phenotypes, with corresponding estimates of prediction performance (2,24).
Figure 3 shows the schematic for running a custom IMP function prediction. A user starts an analysis by specifying an organism and her genes of interest, either manually, from a user-saved gene set, or pre-defined by IMP. Pre-defined gene sets can be from GO or DO, and can include annotations transferred from other organisms by selecting the corresponding checkbox. Figure 3A shows the input for an analysis of five user-provided breast cancer genes. These genes are treated as positive examples for classification, with random negative gene examples selected by IMP for classification. The researcher runs the analysis on IMP's servers using the human functional gene network (Figure 3B). Each gene in the genome is assigned a probability based on its five-fold cross-validated SVM result, and results are sent by email, if provided, or viewed directly on the server though a result-specific URL (Figure 3C). Performance is evaluated as the area under the receiver-operator curve (AUC) and provided with the genome-wide prediction results. As we continue to update IMP's collection of functional networks in the future, the prediction performance of this tool is expected to improve even further, and we encourage biologists to rerun their analyses. With these features, IMP enables biologists to both pose complex biological questions and easily run sophisticated machine-learning tools to help answer them.
Figure 3.
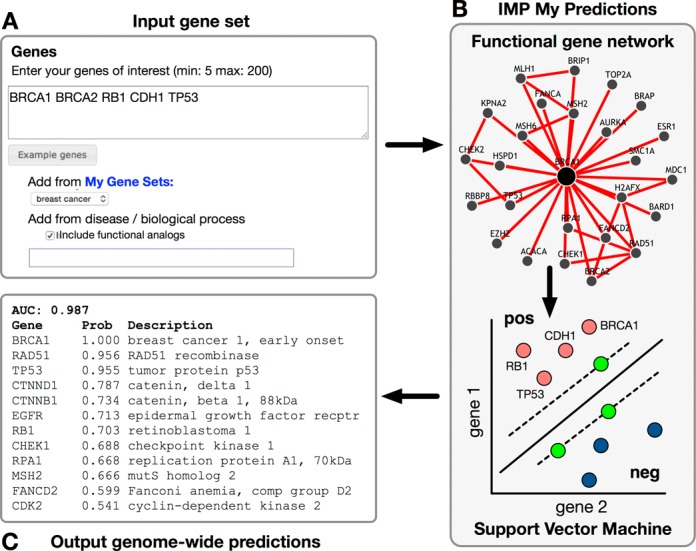
Diagram for submitting custom user predictions. (A) The input form for entering a gene set of interest. Genes can be pasted, selected from a saved gene set, or chosen from a pre-defined set. (B) IMP applies an SVM with the provided gene set as positive examples and predicts additional genome-wide genes likely to be functionally related. (C) The output is a list of genome-wide genes, ranked by their probability of functional relationship with the provided gene set. This result can be emailed to the user or accessed directly on the web server.
SUMMARY
IMP is a flexible, user-friendly web server that serves as an intuitive and accessible resource for molecular biologists who want to leverage heterogeneous biological big data collections to explore predictions of gene function and disease association in human and model organisms. The described updates add substantial value to IMP as a unique resource and suite of analysis tools for biological researchers. In the future, we plan to continue to add additional organisms (Arabidopsis thaliana) and additional data sources for our functional gene networks. We continue to develop additional tools that leverage our cross-organism collection of networks and predictions with the goal of making complex tools and analyses accessible to biological researchers.
FUNDING
National Science Foundation (NSF) CAREER [DBI-0546275]; National Institutes of Health [R01 GM071966, R01 HG005998, T32 HG003284]; National Institute of General Medical Sciences (NIGMS) Center of Excellence [P50 GM071508]. Funding for open access charge: Simons Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ozsolak F., Milos P.M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park C.Y., Wong A.K., Greene C.S., Rowland J., Guan Y., Bongo L.A., Burdine R.D., Troyanskaya O.G. Functional knowledge transfer for high-accuracy prediction of under-studied biological processes. PLoS Comput. Biol. 2013;9:e1002957. doi: 10.1371/journal.pcbi.1002957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong A.K., Park C.Y., Greene C.S., Bongo L.A., Guan Y., Troyanskaya O.G. IMP: a multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res. 2012;40:W484–W490. doi: 10.1093/nar/gks458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zuberi K., Franz M., Rodriguez H., Montojo J., Lopes C.T., Bader G.D., Morris Q. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013;41:W115–W122. doi: 10.1093/nar/gkt533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan Y., Gorenshteyn D., Burmeister M., Wong A.K., Schimenti J.C., Handel M.A., Bult C.J., Hibbs M.A., Troyanskaya O.G. Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput. Biol. 2012;8:e1002694. doi: 10.1371/journal.pcbi.1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huttenhower C., Haley E.M., Hibbs M.A., Dumeaux V., Barrett D.R., Coller H.A., Troyanskaya O.G. Exploring the human genome with functional maps. Genome Res. 2009;19:1093–1106. doi: 10.1101/gr.082214.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., Von Mering C., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmitt T., Ogris C., Sonnhammer E.L.L. FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids ResD. 2014;42:D380–D388. doi: 10.1093/nar/gkt984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chikina M.D., Troyanskaya O.G. Accurate quantification of functional analogy among close homologs. PLoS Comput. Biol. 2011;7:e1001074. doi: 10.1371/journal.pcbi.1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatr-Aryamontri A., Breitkreutz B.-J., Oughtred R., Boucher L., Heinicke S., Chen D., Stark C., Breitkreutz A., Kolas N., O'Donnell L., et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43:D470–D478. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Licata L., Briganti L., Peluso D., Perfetto L., Iannuccelli M., Galeota E., Sacco F., Palma A., Nardozza A.P., Santonico E., et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40:D857–D861. doi: 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerrien S., Aranda B., Breuza L., Bridge A., Broackes-Carter F., Chen C., Duesbury M., Dumousseau M., Feuermann M., Hinz U., et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841–D846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers C.L., Robson D., Wible A., Hibbs M.A., Chiriac C., Theesfeld C.L., Dolinski K., Troyanskaya O.G. Discovery of biological networks from diverse functional genomic data. Genome Biol. 2005;6:R114. doi: 10.1186/gb-2005-6-13-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amberger J.S., Bocchini C.A., Schiettecatte F., Scott A.F., Hamosh A. OMIM.org: online mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku1205. doi:10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kibbe W.A., Arze C., Felix V., Mitraka E., Bolton E., Fu G., Mungall C.J., Binder J.X., Malone J., Vasant D., et al. Disease ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku1011. doi:10.1093/nar/gku1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willis M.S., Rojas M., Li L., Selzman C.H., Tang R.-H., Stansfield W.E., Rodriguez J.E., Glass D.J., Patterson C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am. J. Physiol. Heart Circ. Physiol. 2009;296:H997–H1006. doi: 10.1152/ajpheart.00660.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kedar V., McDonough H., Arya R., Li H.-H., Rockman H.A., Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. U.S.A. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S.N., Czernuszewicz G., Tan Y., Lombardi R., Jin J., Willerson J.T., Marian A.J. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 2012;111:907–919. doi: 10.1161/CIRCRESAHA.112.270207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arber S., Hunter J.J., Ross J., Hongo M., Sansig G., Borg J., Perriard J.C., Chien K.R., Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 22.Hambleton M., Hahn H., Pleger S.T., Kuhn M.C., Klevitsky R., Carr A.N., Kimball T.F., Hewett T.E., Dorn G.W., Koch W.J., et al. Pharmacological- and gene therapy-based inhibition of protein kinase Cα/β enhances cardiac contractility and attenuates heart failure. Circulation. 2006;114:574–582. doi: 10.1161/CIRCULATIONAHA.105.592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molkentin J.D., Robbins J. With great power comes great responsibility: Using mouse genetics to study cardiac hypertrophy and failure. J. Mol. Cell. Cardiol. 2009;46:130–136. doi: 10.1016/j.yjmcc.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan Y., Ackert-Bicknell C.L., Kell B., Troyanskaya O.G., Hibbs M.A. Functional genomics complements quantitative genetics in identifying disease-gene associations. PLoS Comput. Biol. 2010;6:e1000991. doi: 10.1371/journal.pcbi.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]