

Abstract
The metabolism of host cholesterol by Mycobacterium tuberculosis (Mtb) is an important factor for both its virulence and pathogenesis, although how and why cholesterol metabolism is required is not fully understood. Mtb uses a unique set of catabolic enzymes that are homologous to those required for classical β-oxidation of fatty acids but are specific for steroid-derived substrates. Here, we identify and assign the substrate specificities of two of these enzymes, ChsE4-ChsE5 (Rv3504-Rv3505) and ChsE3 (Rv3573c), that carry out cholesterol side chain oxidation in Mtb. Steady-state assays demonstrate that ChsE4-ChsE5 preferentially catalyzes the oxidation of 3-oxo-cholest-4-en-26-oyl CoA in the first cycle of cholesterol side chain β-oxidation that ultimately yields propionyl-CoA, whereas ChsE3 specifically catalyzes the oxidation of 3-oxo-chol-4-en-24-oyl CoA in the second cycle of β-oxidation that generates acetyl-CoA. However, ChsE4-ChsE5 can catalyze the oxidation of 3-oxo-chol-4-en-24-oyl CoA as well as 3-oxo-4-pregnene-20-carboxyl-CoA. The functional redundancy of ChsE4-ChsE5 explains the in vivo phenotype of the igr knockout strain of Mycobacterium tuberculosis; the loss of ChsE1-ChsE2 can be compensated for by ChsE4-ChsE5 during the chronic phase of infection. The X-ray crystallographic structure of ChsE4-ChsE5 was determined to a resolution of 2.0 Å and represents the first high-resolution structure of a heterotetrameric acyl-CoA dehydrogenase (ACAD). Unlike typical homotetrameric ACADs that bind four flavin adenine dinucleotide (FAD) cofactors, ChsE4-ChsE5 binds one FAD at each dimer interface, resulting in only two substrate-binding sites rather than the classical four active sites. A comparison of the ChsE4-ChsE5 substrate-binding site to those of known mammalian ACADs reveals an enlarged binding cavity that accommodates steroid substrates and highlights novel prospects for designing inhibitors against the committed β-oxidation step in the first cycle of cholesterol side chain degradation by Mtb.
Keywords: tuberculosis; cholesterol metabolism; drug target; 3-oxo-23,24-bisnorchol-4-en-22-oyl-CoA; 3-oxo-23,24-bisnorchol-4,17-dien-22-oyl-CoA; 17-hydroxy-3-oxo-23,24-bisnorchol-4-en-22-oyl-CoA
Throughout the course of Mycobacterium tuberculosis (Mtb) infection and tuberculosis (TB) disease, the metabolism of lipids, including steroids such as cholesterol, is preferred by the mycobacteria. Mtb can degrade cholesterol in vitro, even as a sole carbon source, and in vivo the bacteria require cholesterol metabolism for establishing and maintaining chronic infection.1−4 Cholesterol metabolism provides Mtb with a source of acetyl-CoA and propionyl-CoA, which can be utilized for energy production.3,5 In addition, potentially valuable steroid-derived metabolites that might contribute to Mtb’s survival and virulence could be biosynthesized from cholesterol.
Degradation of the branched fatty acid-like side chain of cholesterol is proposed to proceed through β-oxidation,6,7 in a manner that is analogous to fatty acyl-CoA metabolism in human mitochondria and peroxisomes.8 The initiation of β-oxidation requires the conversion of cholesterol to cholest-4-ene-3-one, which is catalyzed by 3β-Hsd,9 followed by the Cyp125-catalyzed oxidation of the C26 terminal methyl10−12 to provide the initial fatty acid that is activated to the CoA thioester by FadD19.13 The complete metabolism of the cholesterol side chain results in a 17-keto steroid intermediate as well as one acetyl-CoA and two propionyl-CoA molecules (Scheme 1).14 In the fatty acid β-oxidation cycle, ACADs typically have the lowest enzymatic throughput by several orders of magnitude and therefore control the overall reaction flux.15 The identification of the specific ACAD enzymes that catalyze each step in the three cycles of β-oxidation is essential to determining the best target to block the generation of propionyl-CoA and acetyl-CoA, and thus energy production, from cholesterol. Moreover, the inhibition of this activity would also potentially block the production of downstream steroid-derived metabolites with the caveat that there are no published reports of 3-oxo-cholest-4-en-26-oyl-CoA (3-OCS-CoA) serving as a substrate for ring-degrading enzymes; however, future studies could potentially demonstrate such activity.
Scheme 1. Cholesterol Side Chain Metabolism in Mycobacterium tuberculosis.

Cyp125 initiates cholesterol side chain degradation.10−12 FadD19 acyl-CoA ligase activates the resultant steroid carboxylic acid through esterification with CoA.13 The steroid side chain is truncated via three cycles of β-oxidation to yield one acetyl-CoA and two propionyl-CoA’s (highlighted in purple) and androstendione. The first step in each β-oxidation cycle is labeled with the acyl-CoA dehydrogenase that catalyzes the oxidation and highlighted in blue text. The bonds undergoing modification are highlighted in red. Additional steps are labeled with specific enzyme names, if known.
Work from our laboratory has demonstrated that the KstR1-regulated igr (intracellular growth) operon encodes the enzymes that catalyze the final β-oxidation cycle in cholesterol side chain degradation that removes the C20 to C22 propionate moiety of the cholesterol side chain (Scheme 1).16−18 ChsE1-ChsE2 catalyzes the dehydrogenation of 3-oxo-4-pregnene-20-carboxyl-CoA (3-OPC-CoA) to 3-oxo-4,17-pregnadiene-20-carboxyl-CoA (3-OPDC-CoA).16,17 ChsH1-ChsH2 catalyzes the hydration of 3-OPDC-CoA to 17-hydroxy-3-oxo-4-pregnene-20-carboxyl-CoA (17-HOPC-CoA).18
We discovered that both ChsE1-ChsE2 and ChsH1-ChsH2 are α2β2 heterotetramers. This α2β2 structural architecture has, thus far, been found only in bacteria known to metabolize sterols. With the insights gained from the study of ChsE1-ChsE2, we further identified additional α2β2 heterotetrameric ACADs encoded in the Mtb genome, including FadE17-FadE18, FadE23-FadE24, FadE26-FadE27, FadE31-FadE32, and FadE31-FadE33.19 All of these ACADs are encoded by genes that reside in single operons and are therefore expressed polycistronically in vivo.19 Furthermore, the expression of all of the α2β2 ACAD enzymes from Mtb are regulated by cholesterol.1
In addition to being induced by cholesterol in Mtb,1 four of the heterotetramer-encoding ACAD genes, fadE26, fadE27, chsE1, and chsE2, as well as a fifth acad gene, fadE34, are in the KstR1 regulon.20 In a related actinobacteria, G. neofelifaecis, these five acad genes are induced by cholesterol but not androstenedione, the sterol metabolite formed after the complete removal of the side chain.21 The demonstrated catalytic activity of ChsE1-ChsE2,16,17 the requirement of a sterol side chain for induction,21 and their repression by KstR120,22 suggest that these five acad genes encode the three ACADs that catalyze the first step of side chain dehydrogenation in the three β-oxidation cycles of cholesterol side chain catabolism (Scheme 1).
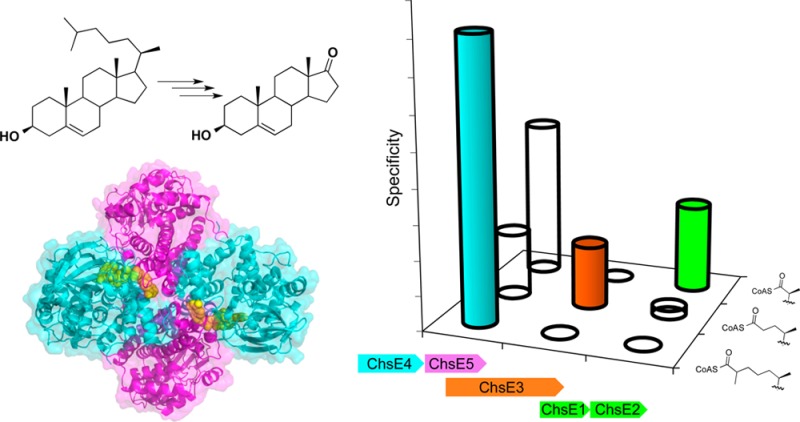
Therefore, we investigated the catalytic activities of fadE26, fadE27, and fadE34 gene products. We employed biophysical characterization, substrate synthesis, and steady-state kinetics to determine which ACADs dehydrogenate the five-carbon and eight-carbon cholesterol side chain metabolic intermediates and the degree to which the substrate specificities overlapped. Our data clearly demonstrate that FadE34 is the ACAD responsible for catalyzing dehydrogenation in the second cycle of cholesterol side chain β-oxidation and that FadE26-FadE27 is the ACAD in the first cycle of β-oxidation. With an established function in cholesterol side chain degradation, we now refer to FadE34 and FadE26-FadE27 as ChsE3 and ChsE4-ChsE5, respectively, to distinguish them from the FadE (fatty acid degradation E) acyl-CoA dehydrogenase subfamily.
The ChsE4-ChsE5 activity profile provides insight into compensatory activities that may contribute to the in vivo phenotype of the igr mutant. The X-ray crystal structure of ChsE4-ChsE5 reveals the apparent evolutionary relationship with the canonical homotetrameric ACADs and key differences between them. The binding-site features of the α2β2 ACAD distinguish it from the mammalian host homotetrameric structure23 and will provide guidance for rational inhibitor design.
Results and Discussion
The α2β2 heterotetrameric acyl-CoA dehydrogenase ChsE1-ChsE2 functions in the last cycle of β-oxidation in cholesterol side chain degradation16,17 (Scheme 1). The chsE1 and chsE2 genes are part of the igr operon, which is regulated by the KstR1 repressor.20,22 We reasoned that the three additional acad genes were regulated by the KstR1 repressor function in a related set of steps during cholesterol metabolism. Two of these genes, chsE4 and chsE5, encode an α2β2 ACAD.19 The third gene, chsE3, encodes a homologue of the very long chain ACAD subfamily. The unusual structural organization of two of these ACADs, the enzymatic activity of ChsE1-ChsE2, and their common regulation led to our hypothesis that the ACADs function in the first and second cycles of sterol side chain β-oxidation. Therefore, we undertook the elucidation of the enzymatic functions and structures of ChsE4-ChsE5 and ChsE3 as well as further substrate specificity testing of ChsE1-ChsE2.
Biophysical Characterization of ChsE4-ChsE5 and ChsE3
ChsE4 and ChsE5 belong to the ACAD family. Typical ACADs catalyze the dehydrogenation of aliphatic acyl-CoAs and are homotetramers with four active sites.23 In contrast, ChsE4-ChsE5 is encoded by two adjacent genes in the Mtb H37Rv genome, chsE4 and chsE5 (Rv3504 and Rv3505, formerly fadE26 and fadE27).19 We previously reported that ChsE4 and ChsE5 form a stable protein with FAD cofactor bound only when the two encoding genes are coexpressed.19 ChsE4-ChsE5 was expressed with an N-terminal His6 tag on ChsE4 in E. coli and purified by IMAC and then further purified by size exclusion chromatography. Additional analysis by sedimentation equilibrium analytical ultracentrifugation (AUC) and LC/UV/MS determined that in solution ChsE4-ChsE5 is an α2β2 heterotetrameric complex that binds two FAD cofactors (Figure S1a).19
ChsE3 (Rv3573c, formerly FadE34) also belongs to the ACAD family. However, the ChsE3 protein sequence is most similar to the very long chain acyl-CoA dehydrogenase (VLCAD) subfamily that forms homodimers rather than homotetramers.23 ChsE3 was expressed as an N-terminal His6-tagged protein in E. coli and purified by IMAC and then further purified by size exclusion chromatography. The UV–vis spectrum of purified ChsE3 showed distinctive absorbance maxima at 370 and 440 nm, indicating the presence of the bound FAD cofactor. Further analysis by sedimentation equilibrium analytical ultracentrifugation (AUC) revealed that ChsE3 forms an α2 homodimer in solution (Figure S1b). There are two FAD cofactors bound per α2 dimer in ChsE3, as expected for a VLCAD subfamily member.
Preparation of the Three Acyl-CoA Metabolic Intermediates of Cholesterol Side Chain β-Oxidation
To test the enzymatic activity of the KstR1-regulated ACADs, we required their putative substrates. Previous work with ChsE1-ChsE2 demonstrated a 5-fold-higher substrate specificity for the ring-intact 3-OPC-CoA ester compared to that for a two-ring indanone CoA ester metabolite.16 Therefore, we undertook the synthesis of the three-carbon, five-carbon, and eight-carbon CoA ester metabolites with the four-ring skeleton. We prepared (3-OPC-CoA) as previously described16 and developed a preparative method for the two additional substrates.
For the preparation of 3-oxo-chol-4-en-24-oyl-CoA (3-OCO-CoA) and 3-oxo-cholest-4-en-26-oyl-CoA (3-OCS-CoA), we protected the A/B ring of stigmasterol as the methyl i-ether prior to ozonolysis of the stigmasterol side chain with a reductive workup and obtained aldehyde 2 (Scheme 2).24 A two-carbon homologation with 2 was performed using the method of Bellassoued and Majidi25 to synthesize enal 7. The final C5 and C8 side chains were constructed using Horner–Wadsworth–Emmons chemistry with aldehydes 2 and 7, respectively.
Scheme 2. Synthesis of (A) 3-Oxo-chol-4-en-24-oyl-CoA (3-OCO-CoA) and (B) 3-Oxo-cholest-4-en-26-oyl-CoA (3-OCS-CoA).

Hydrogenation followed by the deprotection of esters 3 and 8 provided carboxylic acid precursors 5 and 10. Intermediate 9 was obtained as a 1:1 mixture of 25R/25S diastereomers after hydrogenation, and the mixture was subjected to the remaining transformations. These 3β-hydroxy-5-ene acids were readily converted to their corresponding 3-oxo-4-ene acids 6 and 11 using Streptomyces sp. cholesterol oxidase (ChoA).26 The resulting acids were thioesterified using acyl-CoA ligases mtFadD17 and mtFadD19, respectively.27 ChoA and mtFadD19 did not alter the diastereomeric ratio of acid 9, and 3-OCS-CoA was obtained as a 1:1 ratio of 25R/25S thioesters.
ChsE4-ChsE5, ChsE3, and ChsE1-ChsE2 Catalyze the Three Sequential Acyl-CoA Dehydrogenations Required for Cholesterol Side Chain β-Oxidation
In earlier work, we demonstrated that ChsE1-ChsE2 catalyzes the dehydrogenation of 3-OPC-CoA and that the disruption of the igr operon encoding ChsE1-ChsE2 resulted in the accumulation of the analogous methyl ester three-carbon side chain intermediate.16,17 However, the identities of the ACADs that dehydrogenate the five-carbon side chain and eight-carbon side chain cholesterol metabolites were unknown. On the basis of our hypothesis that the KstR1-regulated ACADs are responsible, we determined the steady-state rate constants for ChsE1-ChsE2, ChsE3, and ChsE4-ChsE5 with 3-OPC-CoA,17 3-OCO-CoA, and 3-OCS-CoA as substrates (Table 1). Because the physiological electron-transfer protein is unknown, we used the ferricenium system28 for the oxidation of reduced ACADs.
Table 1. Steady-State Kinetic Parameters for ChsE1-ChsE2, ChsE3, and ChsE4-ChsE5 with the Acyl-CoA Thioesters of the Cholesterol Side Chain Degradation Intermediatesa.
| substrate | KM (μM) | kcat (s–1) | kcat/KM (M–1 s–1) |
|---|---|---|---|
| ChsE1-ChsE2 | |||
| 3-OPC-CoA | 5.3 ± 0.9b | 1.30 ± 0.02b | (2.5 ± 0.5) × 105b |
| 3-OCO-CoA | 6.5 ± 0.9 | 0.16 ± 0.005 | (2.4 ± 0.3) × 104 |
| 3-OCS-CoA | nac | nac | nac |
| ChsE3 | |||
| 3-OPC-CoA | nac | nac | nac |
| 3-OCO-CoA | 28 ± 8 | 5.0 ± 0.5 | (1.8 ± 0.9) × 105 |
| 3-OCS-CoA | nac | nac | nac |
| ChsE4-ChsE5 | |||
| 3-OPC-CoA | 3.3 ± 0.7 | 1.5 ± 0.05 | (4.5 ± 0.9) × 105 |
| 3-OCO-CoA | 2.6 ± 0.4 | 0.48 ± 0.01 | (1.9 ± 0.3) × 105 |
| 3-OCS-CoA | 3.4 ± 1.0d | 2.7 ± 0.4d | (8.1 ± 2.5) × 105 |
| Kiu = 20 ± 2.7 | |||
| octanoyl-CoA | 4.1 ± 0.9 | 0.042 ± 0.002 | (1.0 ± 0.2) × 104 |
Error bars are the standard deviations of global fits to three independent experiments.
Data from ref (17)
No activity.
Inhibition was observed with 3-OCS-CoA. The data were fit by assuming uncompetitive substrate inhibition.
As expected from the metabolism study,16 ChsE1-ChsE2 possessed the highest catalytic activity with 3-OPC-CoA (Figure 1a). Although ChsE1-ChsE2 also catalyzed the dehydrogenation of 3-OCO-CoA, apparent second-order rate constant kcat/Km was 10-fold lower than with 3-OPC-CoA as the substrate. When 3-OCS-CoA was used as a substrate, no catalytic activity was detected. These substrate specificity data further confirmed that ChsE1-ChsE2 is the ACAD responsible for the last cycle of β-oxidation in Mtb cholesterol side chain metabolism.16,17
Figure 1.
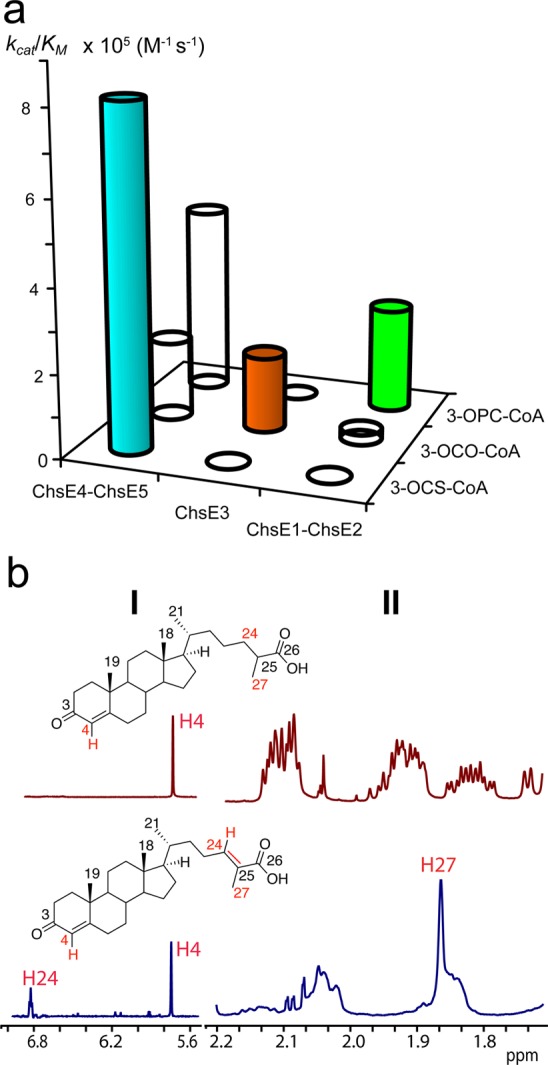
Catalytic specificity for the KstR1-regulated acyl-CoA dehydrogenases and product regio- and stereochemistry for ChsE4-ChsE5. (a) Plot of the catalytic specificity (kcat/KM) of ChsE4-ChsE5, ChsE3, and ChsE1-ChsE2 for the three acyl-CoA metabolic intermediates of cholesterol side chain β-oxidation, 3-OPC-CoA, 3-OCO-CoA, and 3-OCS-CoA, respectively. (b) ChsE4-ChsE5 forms (24E)-3-oxo-cholest-4,24-dien-26-oyl-CoA as determined by the 1H NMR spectra (700 MHz) of substrate precursor 3-oxo-cholest-4-en-26-oic acid and the ChE4-ChsE5 assay product after alkaline hydrolysis of its thioester, top and bottom, respectively. The spectra illustrate the changes in the alkene (I) and methyl (II) regions. The biochemical reaction catalyzed by ChsE4-ChsE5 is shown in Scheme 1.
ChsE3 showed much narrower substrate specificity. The only substrate for which the catalysis of dehydrogenation by ChsE3 was observed is 3-OCO-CoA (Table 1, Figure 1a). No dehydrogenase activity was observed with the three-carbon side chain (3-OPC-CoA) or the eight-carbon side chain (3-OCS-CoA) intermediates as judged by both steady-state spectroscopic assays and MALDI-TOF mass spectrometry of the reaction mixture. The data demonstrate that ChsE3 specifically functions in the second cycle of β-oxidation (Scheme 1).
In contrast, ChsE4-ChsE5 shows the broadest substrate specificity. Although ChsE4-ChsE5 catalyzes the dehydrogenation of all three acyl-CoA substrates (3-OPC-CoA, 3-OCO-CoA, and 3-OCS-CoA), it catalyzes the dehydrogenation of 3-OCS-CoA most efficiently with a kcat/KM that is 2 to 4 times greater than for 3-OCO-CoA or 3-OPC-CoA, respectively (Figure 1a). Although ChsE4-ChsE5 can dehydrogenate octanoyl-CoA, a medium-length fatty acyl ester chain, the kcat/KM is nearly 80-fold lower (Table 1). Notably, ChsE4-ChsE5 is the only ACAD of the three that accepts 3-OCS-CoA as a substrate. This substrate specificity is consistent with an indispensable role in the first cycle of β-oxidation of the eight-carbon side chain.
The analysis of product formation by MALDI-TOF mass spectrometry revealed that the percent substrate conversion remained fixed at a 1:1 ratio of product to substrate, even after overnight incubation at 25 °C and with a 6-fold increase in ChsE4-ChsE5 concentration over that required for turnover (Figure S2c). We reasoned that ChsE4-ChsE5 can dehydrogenate only one diastereomer of 3-OCS-CoA. Most ACADs typically catalyze the dehydrogenation of the (S) α-methyl, acyl-CoA diastereomer.29 In the case of ChsE4-ChsE5, the stereochemistry has not been elucidated. However, the specific dehydrogenation of (25S)-OCS-CoA would be consistent with the stereospecificity of Cyp125, the P450 enzyme that catalyzes the oxidation thatof the C26 methyl of cholest-4-en-3-one two steps earlier in the pathway.11
At high concentrations of 3-OCS-CoA, inhibition was observed. However, no inhibition was observed with either OPC-CoA or 3-OCO-CoA. We assume that inhibition is due to the presence of unreactive 3-OCS-CoA diastereomer that binds to ChsE4-ChsE5 nonproductively. Therefore, the steady-state kinetic data were fit to a modified Michaelis–Menten equation that took uncompetitive substrate inhibition into account in order to estimate kcat and KM.
ChsE4-ChsE5 Forms (24E)-3-Oxo-cholest-4,24-dien-26-oyl-CoA
The ChsE4-ChsE5 reaction product was subjected to alkaline hydrolysis, and the free acids were purified by extraction. A comparison of 1H spectra of 3-oxo-cholest-4-en-26-oic acid 11 and the hydrolyzed ChsE4-ChsE5 reaction product (Figure 1b) highlights the appearance of a new proton resonance in the product spectrum alkene region at 6.85 ppm, consistent with unsaturation at C24–C25 (Figure 1b). As expected, only half of the reaction mixture was converted to product, and the C24 alkene hydrogen resonance integrates to 0.5 hydrogen. A comparison of the H24 chemical shift to chemical shifts calculated for E and Z alkene resonances of α,β-unsaturated carboxylic acids30 reveals that the E alkene is formed. Analogous NMR experiments with the 3-OCO-CoA reaction product revealed that the (22E)-3-oxo-chol-4,22-dien-24-oyl-CoA product was formed (Figure S3).
In addition, a new methyl resonance that corresponds to the C27 methyl of the α,β-unsaturated acid appears as a singlet at 1.85 ppm (Figure 1b). If C25–C27 dehydrogenation had occurred, then two new alkene protons would have replaced the C27 methyl in the product spectrum, and there would be no methyl resonance at 1.85 ppm. We conclude that ChsE4-ChsE5 forms the thermodynamically favored (E)-tetrasubstituted alkene product (24E)-3-oxo-cholest-4,24-dien-26-oyl-CoA.
First Molecular Structure of an α2β2 Heterotetrameric Acyl-CoA Dehydrogenase in Any Kingdom of Life
Because there is no molecular structure of an α2β2 heterotetrameric ACAD, we undertook the structure determination of ChsE4-ChsE5 using X-ray crystallography. Crystals were obtained at pH 7.0 using the hanging drop vapor diffusion method. The crystal structure was determined using single-wavelength anomalous dispersion (SAD) phasing of the selenomethionine (SeMet) substituted protein. The asymmetric unit contains one α2β2 heterotetramer with two ChsE4 chains and two ChsE5 chains, and the initial model was refined to a resolution of 2.0 Å (Table 2).
Table 2. Data Collection and Refinement Statistics.
| crystal | ChsE4-ChsE5/FADH2a |
|---|---|
| space group | C 1 2 1 |
| cell dimensions | |
| a, b, c (Å) | 185.7, 108.1, 82.0 |
| α, β, γ (deg) | 90.0, 93.0, 90.0 |
| data collectionb | |
|---|---|
| resolution (Å) | 46.4–2.0 (2.00–1.99) |
| wavelength (Å) | 0.9792 |
| reflections | |
| observed | 739 658 (7336) |
| unique | 109 767 (1104) |
| Rmergec | 0.076 (0.624) |
| Rmeasd | 0.082 (0.700) |
| Ranome | 0.057 (0.588) |
| Rpimf | 0.031 (0.262) |
| CC1/2g | 0.999 (0.939) |
| I/σI | 19.9 (2.9) |
| completeness (%) | 99.9 (99.8) |
| multiplicity | 6.7 (6.6) |
| Wilson B | 28.9 |
| refinementb | |
|---|---|
| resolution (Å) | 46.4–2.0 (2.05–1.99) |
| no. reflections | 104 247 (7657) |
| Rwork/Rfree | 0.1508/0.1862 |
| CC*g | 1.000 (0.984) |
| no. atoms | |
| protein | 11 396 |
| FADH2 | 106 |
| solvent | 899 |
| B factors | |
| protein | 36.6 |
| FADH2 | 32.5 |
| solvent | 42.5 |
| RMS deviations | |
| bond lengths (Å) | 0.017 |
| bond angles (deg) | 0.84 |
| Ramachandran | |
| favored (%) | 98 |
| allowed (%) | 2 |
| outliers (%) | 0 |
| PDB ID | 4X28 |
See Figure S5.
Values in parentheses are for the highest-resolution shell.
The merging R factor describes the deviation of an individual intensity measurement from the mean value of all of its symmetry-equivalent reflections.
The redundancy-independent merging R factor, Rrim or Rmeas, indicates the precision of an individual intensity measurement independently of the multiplicity of that measurement.
The precision-indicating merging R factor, Rpim, describes the precision of the averaged intensity measurement.
The anomalous R factor quantifies the relative differences in Friedel-related reflections.
CC1/2 and CC* are statistics for assessing the effective resolution limits and quality of diffraction data in the context of a refined model.31
The complete tetramer is a dimer of heterodimers in a tetrahedral arrangement (Figure 2a). Each heterodimer contains a single FAD binding site. The binding site is at the heterodimer interface and is composed of residues from both ChsE4 and ChsE5, as previously predicted17,19 (Figure 2a,b). The molecular envelope of ChsE4-ChsE5 in solution, determined from small-angle X-ray scattering, agrees well with the shape of the tetrameric crystal structure with a χ value of 2.8 fitting from CRYSOL, thus confirming that the observed tetrameric architecture is the native solution state and not a result of crystal packing (Figure S4).
Figure 2.
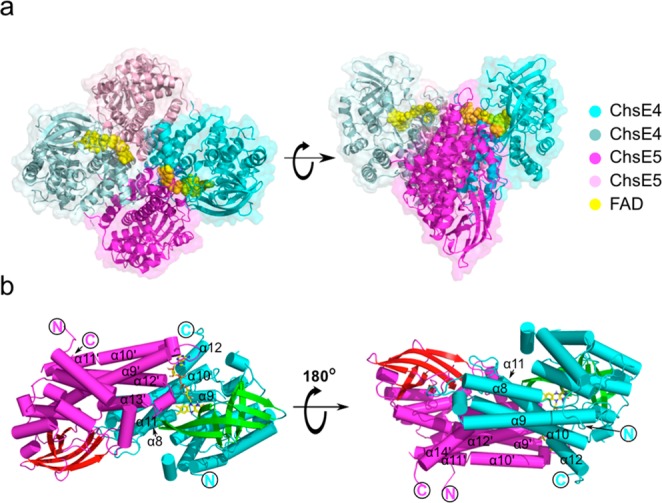
Overall atomic picture of ChsE4-ChsE5. (a) The biological functional unit is an α2β2 heterotetramer with two ChsE4 chains and two ChsE5 chains. The tetramer has two flavin adenine dinucleotide (FAD) binding sites, and two FADs are bound. The surface representation of the structure corresponds to the chain color. (b) A ChsE4-ChsE5 αβ heterodimer is shown as a cylindrical cartoon and colored by the secondary structure. FAD binds to the interface of ChsE4 and ChsE5. The structure on the right is a 180° rotation around the X axis of the structure on the left.
Without any substrate bound to the ChsE4-ChsE5 structure and therefore no electron acceptor present, reduced FAD (a mixture of FAD•– and FADH2) was observed in the ChsE4-ChsE5 structure. A simulated annealing Fo-Fc omit electron density map of the bound FAD molecules in ChsE4-ChsE5 was calculated and showed an obvious bend of 154–156° in the electron density for the isoalloxazine moiety, instead of the conjugated flat electron density map that would be expected for the oxidized form. Using correlated optical spectroscopy and X-ray diffraction data collection, we determined that the FAD is reduced by X-ray irradiation (Figure S5), and we refined the structure with FADH2.
The overall fold of both ChsE4 and ChsE5 is similar to that of typical ACADs, the quintessential example being medium-chain acyl-CoA dehydrogenase (MCAD).23 ChsE4 and ChsE5 both contain a typical N-terminal ACAD domain, a typical C-terminal ACAD domain, and a middle β-barrel domain, which are comparable in size (Figure S6). The N-terminal domains, consisting of mainly α-helices, lie on the surface of the tetramer (Figure 3b). The middle β-barrel domains of ChsE4 and ChsE5 are composed of two orthogonal β-sheets. The middle domain of ChsE4 is partially buried inside the tetramer and interacts with the FAD molecules. In contrast, the middle domains of ChsE5 are located completely on the surface (Figure 3a,b). The C-terminal domains are primarily composed of α-helices and form a four-helix up and down bundle. The C-terminal domains pack against each other to form the central core of the whole tetrahedral assembly.
Figure 3.

Topology scheme for the ChsE4-ChsE5 α2β2 heterotetramer and organization of ACAD domains relative to the tetramer and monomer–monomer interfaces. (a) Topology scheme for the ChsE4-ChsE5 heterotetramer. The functional heterodimers are circled in red boxes with FAD bound at the interface of ChsE4 and ChsE5; the isoalloxazine ring of FAD is highlighted in the yellow circle. (b) N-terminal domains of both ChsE4 and ChsE5 are on the surface of the tetramer; C-terminal domains of both ChsE4 and ChsE5 are at the monomer–monomer interfaces and form the core of the tetramer. Middle β-barrel domains from ChsE5 are on the surface of the tetramer, whereas middle β-barrel domains from ChsE4 are partially exposed to the solvent and the other face contacts FAD. (c) The ChsE4 and ChsE5 interface forms the functional heterodimer with an FAD bound at the interface. (d) The ChsE4 and ChsE5 interface forms a pseudoheterodimer without a bound FAD. (d) 90° rotation around the Y axis of (c).
ChsE4-ChsE5 Monomer–Monomer Interface
Because of the divergent sequences and resultant asymmetry of ChsE4 and ChsE5, two different monomer–monomer interfaces are present in the heterotetrameric assembly. Interactions involving α10, α11, and α12 from ChsE4 with α11′, α12′, and α13′ from ChsE5 comprise a functional ChE4-ChsE5 heterodimer with one FAD cofactor bound (Figure 3c). Meanwhile, α8 and α9 from the same ChsE4 interact with α9′, α10′, α11′, and α13′ from the other ChsE5 chain in the tetramer to form a pseudoheterodimer without a bound FAD cofactor (Figure 3d).
ChsE4 and ChsE5 Are Structurally Similar and Complementary
As previously discussed (vide supra), one ChsE4 chain and one ChsE5 chain are in intimate contact to form an FAD binding site (Figure 2b). The majority of the contacts are between the C-terminal domain helices (Figures 3c and 4a,b). Specifically, the isoalloxazine ring lies in the cavity composed of the hydrophobic faces of the ChsE4 C-terminal domain and middle β-barrel domain (Figures 3a and 4b). The diphosphate and ribose sugar are bonded to residues from helices of the C-terminal domain of ChsE5. Just as in a canonical homotetramer,23 half of the FAD binding residues are contributed by the ChsE4 protomer and the other half are from the ChsE5 protomer, together forming an intact FAD binding site (Figure 4a,b).
Figure 4.
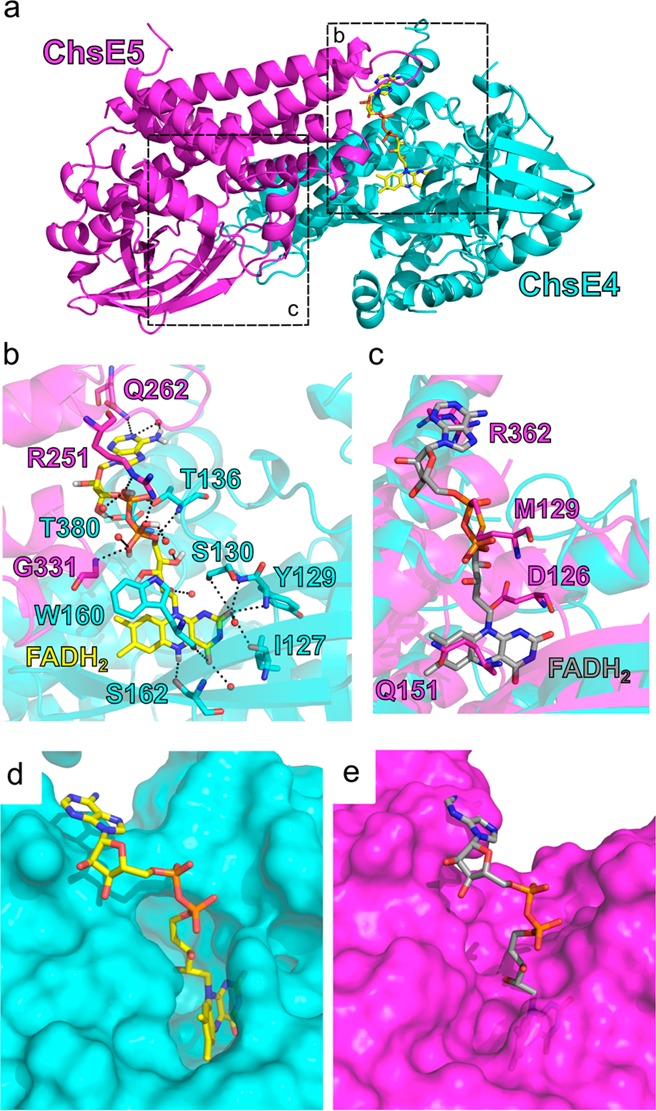
FAD binding sites. (a) There is only one FAD cofactor per ChsE4-ChsE5 dimer. The FAD cofactor binds at the interface of the ChsE4 and ChsE5 chains. The actual FAD binding site is in box b, and the nonfunctional FAD binding region is in box c. (b) The detailed hydrogen bonding network that stabilizes bound FAD is shown. FAD is colored yellow. Water molecules are shown as red balls. Hydrogen bonding interactions are shown as black dashes. Residues that interact with FAD are labeled. The isoalloxazine ring is located inside ChsE4, and both ChsE4 and ChsE5 stabilize adenine. (c) The region in ChsE5 equivalent to the FAD binding site in ChsE4 is shown after superimposing ChsE5 onto ChsE4/FAD. The FAD molecule bound to ChsE4 is shown in gray, and residues from ChsE5 that would clash with FAD are shown and labeled. (d) Surface representation of the actual FAD binding site in ChsE4. Protein chains are shown on the surface, and the FAD molecule is colored in yellow and represented by sticks. (e) Surface representation of the nonfunctional FAD binding region in ChsE5. The FAD molecule is colored in gray.
In distinct contrast to the ChsE4 C-terminal domain, the ChsE5 C-terminal domain does not bind an isoalloxazine ring. The lack of bound FAD in the crystal structure is consistent with the stoichiometry of FAD/ChsE4-ChsE5 binding in solution. The superposition of the ChsE4 domain with the ChsE5 domain revealed that the analogous region of ChsE5 is unsuitable for binding an isoalloxazine moiety (Figure 4c,d). The side chains of ChsE5 residues Arg362, Met129, Asp126, and Gln151 fill the FAD site and thus block the FAD binding (Figure 4c), which is consistent with the surface representation in which ChsE5 conflicts with the conserved FAD binding (Figure 4e). In ChsE4, homologous residues Gln262, Thr136, Ser130, and Ser162 line a larger cavity and are positioned to form hydrogen bonds with FAD (Figure 4b).
Two, Not Four, Substrate Binding Pockets Are Present in the ChsE4-ChsE5 Tetramer
The acyl-CoA substrate-binding pocket is necessarily proximal to the bound FAD cofactor. In typical ACADs (for example, the well-characterized MCAD), there are four acyl-CoA binding pockets adjacent to four FAD cofactors.23,32 Therefore, we analyzed whether the presence of only two FAD binding pockets in the ChsE4-ChsE5 tetramer was correlated with a commensurate reduction in the number of substrate binding pockets in the heterotetramer.
We superimposed the homodimer of MCAD (PDB: 3MDE) with the ChsE4-ChsE5 heterodimer and examined the position of FAD/octanoyl-CoA from MCAD in the ChsE4-ChsE5 heterodimer (Figure 5a). As expected, one MCAD FAD perfectly overlays the FAD from ChsE4-ChsE5 (Figure 5b), and the other MCAD FAD exhibits steric clashes with residues from ChsE5 as seen in the ChsE4-ChsE5 superposition (Figure 4c). This superposition further confirms that ChsE5 cannot accommodate the isoalloxazine moiety.
Figure 5.
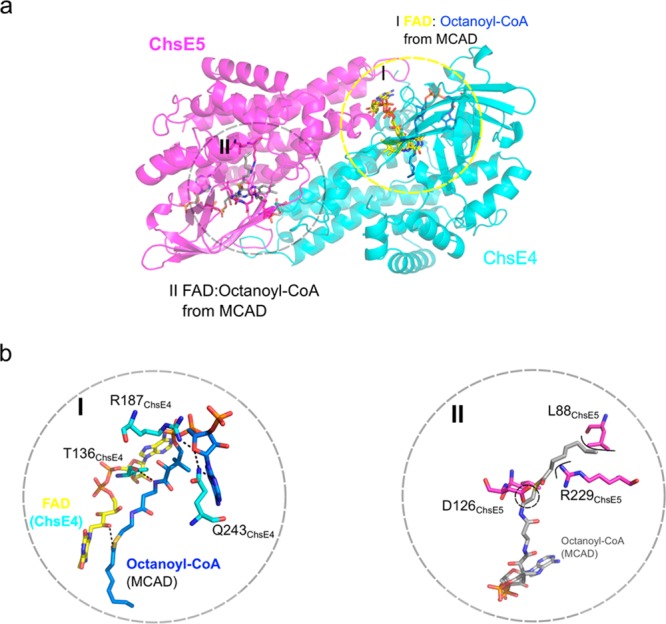
Acyl-CoA binding site. (a) The ChsE4-ChsE5 heterodimer was superimposed onto the MCAD homodimer (PDB: 3MDE) with two FAD/acyl-CoA binding sites. The ChsE4-ChsE5 heterodimer is shown with the two sets of FAD/acyl-CoAs from MCAD. One of the FAD cofactors completely overlays the FAD from ChsE4, circled in I and colored in yellow, and the octanoyl-CoA from MCAD is shown in blue. The other FAD/octanoyl-CoA binding site from MCAD is circled in II and shown in gray. (b) Highly conserved residues T136, R187, and Q243 together with the FAD from ChsE4 interact with the CoA moiety in binding site I. Residues L88, D126, and R229 from ChsE5 would clash with the octanoyl-CoA, shown in circle II. In addition, highly conserved CoA interaction residues T, R, and Q are not conserved in binding site II.
In all the holo-ACAD structures with the acyl-CoA ligand bound, the coenzyme A moiety is exposed to solvent. The bound conformation is stabilized by hydrogen bonding interactions with the protein, and the aliphatic chains extend deep into a hydrophobic protein pocket from which solvent is excluded. Octanoyl-CoA from the MCAD structure superimposes directly into the ChsE4 hydrophobic pocket without any steric clashes. The CoA moiety is stabilized by hydrogen bonds with ChsE4 residues Thr136, Arg187, and Gln243. These residues are highly conserved among ACAD CoA binding sites (Figure 5b).17
The second MCAD octanoyl-CoA does not fit into ChsE5. Specifically, residues Leu88, Asp126, and Arg229 project into the proposed binding tunnel and sterically clash with the docked aliphatic chain of octanoyl-CoA (Figure 5b). Acidic and basic side chains further disfavor binding hydrophobic substrates. Moreover, the highly conserved residues that would ordinarily stabilize CoA binding are absent in ChsE5. Thus, the ChsE5 sequence and therefore structure diverges from the typical ACAD sequence and structure such that it does not possess isoalloxazine or acyl-CoA binding pockets and does not have a catalytic site or an obvious metabolite binding site.
We then examined the superposition of ChsE4 and ChsE5 to further characterize the differences in binding sites in the two protomers. In addition to the presence of large polar residues that occlude the region of ChsE5 that typically would be an acyl-CoA binding tunnel, the superimposed structures of ChsE4 and ChsE5 reveal a conformational shift relative to ChsE4. In ChsE5, secondary structures α11′, α12′, α13′, β6′, β7′, and β3′ are all shifted relative to ChsE4, and instead of comprising a very flexible loop as in ChsE4, α7′ in ChsE5 occupies a relatively large space in what would have been the binding pocket. These shifts further reduce the ChsE5 binding cleft (Figure 6a).
Figure 6.
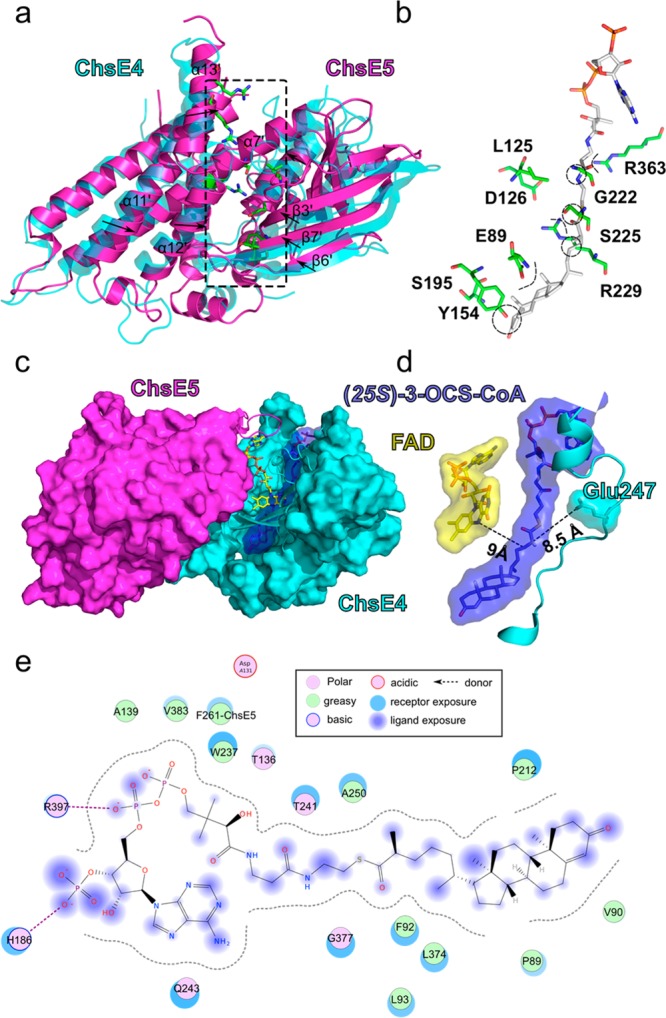
Comparison of ChsE4 and ChsE5 for steroid acyl-CoA binding; model of OCS-CoA bound to the ChsE4-ChsE5 heterodimer. (a) ChsE5 was superimposed onto ChsE4, and the RMSD value is 2.958 Å with 887 α-carbons aligned. The ChsE5 secondary structure that corresponds to the binding tunnel in ChsE4 is shifted into the tunnel relative to ChsE4; the shifting directions are shown by black arrows. The residues that surround the binding tunnel in ChsE5 are shown as sticks and are colored by atom type. (b) Enlarged region of the ChsE5 residues that align with the ChsE4 binding tunnel. These residues have polar, basic, or acidic side chains and clash with the docked substrate (colored by atom) steroid and pantotheine chain. (c) (25S)-OCS-CoA docked into ChsE4 and sandwiched between FAD and the active-site base. For clarity, half of the FAD binding tunnel is represented as a surface colored by a chain, and the other half of the tunnel is drawn as a cartoon. (d) Orientation and proximity of FAD (yellow), the docked 3-OCS-CoA (blue), and the active-site base (light blue). The distances between Glu247 and C25 in 3-OCS-CoA and between C24 in 3-OCS-CoA and N5 in FAD are indicated as dashed lines and labeled. (e) Scheme of the interactions between the ChsE4-ChsE5 heterodimer and (25S)-3-OCS-COA.
Substrate-Binding Pocket of ChsE4-ChsE5 Can Accommodate Bulky Steroid Substrates
Next, we docked substrate (25S)-3-OCS-CoA into the ChsE4-ChsE5 binding pockets. Steric clashes precluded docking into the ChsE5 pocket, again suggesting that ChsE5 does not possess the ability to bind steroid substrates because of the reduced size of the cleft, which is filled with hydrophilic residues (Figure 6b). Conversely, the 3-OCS-CoA substrate could be readily docked into the ChsE4 site, demonstrating that the ChsE4 binding site is sufficiently large to accommodate a steroid structure.
The 3-OCS-CoA steroid side chain aligns between the FAD cofactor and the putative active-site base, Glu247. In homotetrameric ACAD structures, the active-site base is located on an α-helix in close proximity to the proton to be abstracted. In contrast, Glu247 resides on a random coil that interrupts this α-helix (Figure 6c,d). Glu247 is too far from the docked structure for proton transfer to occur, suggesting that a conformational change occurs upon substrate binding. Two hydrogen-bonding interactions are formed between the phosphate group of the CoA molecule and His175 and Arg381. NH3″ interacts with Thr130, and the thioester carbonyl group is stabilized by the backbone amide of Gly362 through hydrogen bonding. Except for a few polar interactions, 3-OCS-CoA is mainly surrounded by hydrophobic and aromatic residues (Figure 6e).
Glu247 from ChsE4 Is the Active-Site Base Required for Dehydrogenation
Consistent with the substrate docking results described above, the alignment of the primary amino acid sequences of ChsE4 and ChsE5 against nine well-characterized human ACADs revealed that ChsE4 possesses the typical catalytic general base glutamate in the conserved position but ChsE5 does not.19 To confirm experimentally that ChsE4-Glu247 is the active-site base, we mutated ChsE4-Glu247 to Ala.
Purified mutant ChsE4E247A-ChsE5 had a characteristic FAD absorbance in the UV–visible spectrum (Figure S2b). ChsE4E247A-ChsE5 was characterized by analytical gel filtration and was found to have the same elution profile as wild-type ChsE4-ChsE5, indicating that ChsE4E247A-ChsE5 still forms a stable α2β2 heterotetramer in solution. The activity of ChsE4E247A-ChsE5 was assayed under the same conditions as for ChsE4-ChsE5. However, no detectable activity was observed even at very high concentrations of enzyme (5 μM) and 3-OCS-CoA substrate (100 μM). Furthermore, no product formation was observed by MALDI-TOF spectrometry (Figure S2c). These results indicate that Glu247 in ChsE4 is the active-site general base required to afford a dehydrogenated product.
Substrate Binding Tunnel Comparison across ACADs
The ability of ChsE4-ChsE5 to bind and catalyze the dehydrogenation of steroid acyl-CoA substrates led us to analyze further the structural details of its substrate-binding tunnel. The substrate binding sites of human isovaleryl-CoA dehydrogenase (i3VD), Mycobacterium smegmatis FadE13, pig medium chain acyl-CoA dehydrogenase (MCAD), rat short chain acyl-CoA dehydrogenase (SCAD), and Megasphaera elsdenii butyryl-CoA dehydrogenase (BCAD) were compared using Caver.33 All of the binding sites are located in the enzyme core (Figure 7a). Of all of the binding sites, ChsE4-ChsE5 has the longest tunnel-like binding site (Figure 7a).
Figure 7.

Substrate binding site comparison across species. (a) The structures of ChsE4, human isovaleryl-CoA dehydrogenase (i3VD), pig medium chain acyl-CoA dehydrogenase (MCAD), Mycobacterium smegmatis FadE13, rat short chain acyl-CoA dehydrogenase (SCAD), and Megasphaera elsdenii butyryl-CoA dehydrogenase (BCAD) are superimposed, and their substrate binding sites have been identified by Caver.28 The substrate binding sites are shown as transparent surfaces. The RMSD value between ChsE4 and i3VD is 2.030 Å with 975 α-carbons aligned; the RMSD value between ChsE4 and 3OIB is 1.718 Å with 831 α-carbons aligned; the RMSD value between ChsE4 and 3MDE is 2.362 Å with 942 α-carbons aligned; the RMSD value between ChsE4 and 1JQI is 2.714 Å with 941 α-carbons aligned; the RMSD value between ChsE4 and 1BUC is 2.086 Å with 870 α-carbons aligned. (b) Secondary structure sequence alignment of ChsE4, i3VD, MCAD, SCAD, BCAD, and FadE13. ChsE4 is colored in cyan, and the other secondary structure cartoons are colored in gray. Yellow highlighted residues are identical; black and gray highlighted residues are very similar.
A structure-based sequence alignment of the homologues revealed differences that reflect the substrate specificity. Even though all of the ACADs have similar (homologous) structures, the distinctive loops between α4 and α5, α7, and α8 give ChsE4 the ability to bind bulkier substrates by proving both more flexibility and a larger amount of space (Figure 7b), supporting its unique biological function to degrade bulky cholesterol in Mtb.
Substrate Promiscuity of ChsE4-ChsE5 Explains the igr Phenotype
ChsE4-ChsE5 is the only ACAD in the KstR1 regulon that can dehydrogenate 3-OCS-CoA. Thus, ChsE4-ChsE5 controls metabolic flux into cholesterol side chain β-oxidation. However, ChsE4-ChsE5 also catalyzes the dehydrogenation of 3-OPC-CoA and 3-OCO-CoA, albeit at somewhat slower rates. The substrate specificity overlap of ChsE4-ChsE5 with ChsE1-ChsE2 and ChsE3 (Table 1) requires that compensating enzyme activities must be considered in the evaluation of ACAD gene knockout phenotypes in vivo and in vitro.
The in vivo and in vitro phenotypes of the igr knockout have been evaluated.4,16,34 Notably, the initial growth of H37Rv:Δigr in the mouse model of infection is attenuated.34 The attenuation has been attributed to the inability of the Δigr mutant to metabolize cholesterol fully.4 A 3-OPC-CoA-related metabolite accumulates in Δigr mutant cultures grown in the presence of cholesterol for 2 weeks, consistent with the loss of igr-encoded ChsE1-ChsE2 activity.16 However, 24 weeks after infection in vivo, Δigr mutant bacterial counts in the lung reach wild-type levels.34
Taking these results together, we infer that in the early stages of infection the catalytic activity of ChsE4-ChsE5 with 3-OPC-CoA is insufficient to overcome the loss of ChsE1-ChsE2 but that in later stages ChsE4-ChsE5 is able to compensate for the deficiency. This change may be attributed to a shift in metabolite concentrations. Although we do not know the concentration of the steroid CoA metabolites in vivo, typically intracellular substrate concentrations of metabolites are close to or higher than Km of the respective catalyzing enzyme.35 We propose that during the initial stages of infection 3-OCS-CoA levels are sufficiently high that ChsE1-ChsE2 is required for the metabolism of 3-OPC-CoA. As infection reaches the chronic phase, the supply of 3-OCS-CoA becomes depleted, 3-OPC-CoA accumulates, and as the substrate specificities suggest (Table 1, Figure 1a), ChsE4-ChsE5 can compensate for the loss of ChsE1-ChsE2. We hypothesize that similarly there are enoyl-CoA hydratases and retroaldolases that compensate for the loss of the igr operon once the dehydrogenation of 3-OPC-CoA has been accomplished.
Conclusions
The substrate specificities of the KstR1-regulated ACADs for sterol side chain degradation in Mtb have been elucidated. Only ChsE4-ChsE5 catalyzes the dehydrogenation of the eight-carbon side chain steroid intermediate, 3-OCS-CoA, which is required for the first cycle of steroid side chain β-oxidation. ChsE3 catalyzes only the dehydrogenation of the five-carbon steroid side chain intermediate, 3-OCO-CoA, and as previously demonstrated,16,17 ChsE1-ChsE2 specifically catalyzes the dehydrogenation of the three-carbon steroid chain intermediate, 3-OPC-CoA.
ChsE4-ChsE5 has the broadest substrate specificity. Although the steady-state kinetics of ChsE4-ChsE5 show a preference for the dehydrogenation of an eight-carbon side chain substrate, both the five-carbon and three-carbon substrates are dehydrogenated at rates comparable to the catalysis of their dehydrogenation by ChsE1-ChsE2 and ChsE3. These secondary activities allow kinetic compensation for the loss of the second (ChsE3) or third (ChsE1-ChsE2) ACAD in steroid side chain β-oxidation. ChsE4-ChsE5 preferentially dehydrogenates steroid substrates, as demonstrated by the 80-fold-lower apparent second-order rate constant for octanoyl-CoA.
The structure of ChsE4-ChsE5 serves as a starting point for the development of specific inhibitors for this subfamily of structurally distinct ACADs. The ChsE4-ChsE5 substrate-binding site is much larger than those seen in host ACADs. Although the α2β2 heterotetramer structure is highly conserved in mycobacterial and proteobacterial species, it is distantly related to typical ACADs, and no known human orthologs exist.17,19,36 Appropriately designed inhibitors that prevent Mtb from generating downstream cholesterol catabolites acetyl-CoA and propionyl-CoA by blocking the first step of cholesterol side chain β-oxidation will allow direct investigation into the role this pathway plays in Mtb persistence and survival in vivo.
Methods
Materials, Strains, Media, and General Methods
Ferricenium hexafluorophosphate was purchased from Sigma-Aldrich (St. Louis, MO). Stigmasterol and coenzyme A were purchased from MP Biomedicals (Solon, Ohio). Isopropyl β-d-1-thiogalactopyranoside (IPTG) was purchased from Denville Scientific (Metuchen, NJ). Tryptone, HEPES, and TRIS were purchased from Fisher Scientific (Pittsburgh, PA). Kanamycin was purchased from IBI Scientific (Peosta, IA). Yeast extract was purchased from Research Products International Co. (Mount Prospect, IL). iProof DNA polymerase was purchased from Bio-Rad (Hercules, CA). Restriction endonucleases, T4 DNA ligase, T4 polynucleotide kinase, and the protein ladder were purchased from New England Biolabs (Beverly, MA). HisTrap FF columns and Superdex 200 HiLoad 16/60 and 10/300 GL columns were purchased from GE Healthcare Biosciences Corp. (Piscataway, NJ). Oligonucleotides were purchased from IDT Inc. (Coralville, IA). The total genomic DNA of M. tuberculosis H37Rv was obtained from the TB Research Materials Facility at Colorado State University (Fort Collins, CO) (NIAD NO1-AI40091). MALDI mass spectra were acquired on a Bruker Autoflex II TOF/TOF. Big dye DNA sequencing (Applied Biosystems, Foster City, CA; performed by the Stony Brook University Sequencing Facility) was used to verify the coding sequence of the expression plasmids. BL21 (DE3) E. coli was obtained from Bio-Rad. Protein expression 2 × YT media is composed of 16 g of tryptone, 10 g of yeast extract, and 5 g of NaCl per liter. Cell disruption was performed using a Constant Systems, Inc. TS series benchtop instrument (Kennesaw, GA). Buffer A: 20 mM Tris-HCl buffer pH 8.0, supplemented with 300 mM NaCl and 10 mM imidazole. Buffer B: 20 mM Tris-HCl buffer pH 8.0, supplemented with 300 mM NaCl and 500 mM imidazole. Buffer C: 50 mM Tris-HCl buffer pH 8.0, supplemented with 200 mM NaCl. Primary crystallization screens were from Hampton Research. LC/MS analysis and ESI-MS were conducted on a Waters UPLC/MS instrument with a diode array and SQD detectors. UV–visible spectra were acquired on a Shimadzu UV2550 UV/visible light spectrophotometer. NMR data were acquired on 400, 500, and 700 MHz Bruker spectrometers. Chemical shifts are denoted in ppm (δ) and calibrated from residual undeuterated solvents. Assigned resonances are reported, and the full spectra are provided in the Supporting Information. Flash chromatography was carried out on a CombiFlash Rf 200 (Teledyne Isco, Lincoln, NE) system.
(E)-t-Butyl 6β-Methoxy-3α,5-cyclo-5α-chol-22-ene-24-ate (3)
To a cooled (0 °C, on ice) solution of sodium hydride (32 mg, 1.3 mmol) in 1 mL of THF was added tert-butyl diethyl phosphonoacetate (328 mg, 1.3 mmol) dropwise. After the mixture was stirred for 5 min, a solution of 6β-methoxy-3α,5-cyclo-5α-pregnane-20-carboxaldehyde 2(24) (227 mg, 0.66 mmol) in 5 mL of THF was added. The resultant mixture was allowed to warm to 25 °C and stirred for 12 h. THF was evaporated, and the product was purified by flash chromatography with EtOAc and hexane (1:19) to yield (E)-t-butyl 6β-methoxy-3α,5-cyclo-5α-chol-22-ene-24-ate (3) (200 mg, 68%). 1H NMR (500 MHz, CDCl3) δ 6.72 (dd, J = 15.5, 8.9 Hz, 1H), 5.64 (d, J = 14.9 Hz, 1H), 3.31 (s, 3H), 2.75 (t, J = 2.7 Hz, 1H), 1.46 (s, 9H), 1.06 (d, J = 6.6 Hz, 3H), 1.01 (s, 3H), 0.73 (s, 3H), 0.68–0.59 (m, 1H), 0.42 (dd, J = 8.0, 5.1 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ 166.5 (C24), 153.6 (C22), 120.6 (C23), 82.4 (OMe), 79.9 (OtBu).
t-Butyl 6β-Methoxy-3α,5-cyclo-5α-chol-24-ate (4)
A solution of 3 (200 mg, 0.45 mmol) in 5 mL of MeOH containing Pd/C (10% w/w, 50 mg) was bubbled with H2 for 1 h. The solution was filtered through a pad of Celite and concentrated in vacuo to yield t-butyl 6β-methoxy-3α,5-cyclo-5α-chol-24-ate (4) (192 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 3.32 (s, 3H), 2.76 (t, J = 2.8 Hz, 1H), 1.44 (s, 9H), 1.02 (s, 3H), 0.91 (d, J = 6.5 Hz, 4H), 0.71 (s, 3H), 0.64 (t, J = 4.4 Hz, 1H), 0.42 (dd, J = 8.0, 5.1 Hz, 1H). 13C NMR (400 MHz, CDCl3) δ 173.7 (C24), 82.4 (OMe), 79.9 (OtBu).
3-Hydroxy-chol-5-ene-24-oic acid (5)
Ester 4 (192 mg, 0.44 mmol) was dissolved in 1 mL of trifluoroacetic acid (TFA), and the resultant mixture was stirred at 25 °C for 12 h. TFA was evaporated, and the remaining solid was dissolved in 2 mL of 1 N KOH/methanol solution. The solution was stirred for 1 h and acidified with 1 N HCl to pH 2. The white precipitate was extracted with EtOAc (50 mL × 3). The EtOAc layer was combined, dried over Na2SO4, and concentrated in vacuo. The product was purified by flash chromatography with EtOAc and hexane (1:4) to yield 3-hydroxy-chol-5-ene-24-oic acid (5) (48 mg, 30%). 1H NMR (500 MHz, CD3OD) δ 5.38–5.37 (m, 1H), 3.45–3.40 (m, 1H), 1.05 (s, 3H), 0.99 (d, J = 6.6 Hz, 3H), 0.76 (s, 3H). 13C NMR (500 MHz, CD3OD) δ 176.8 (C24), 140.8 (C5), 121.0 (C6), 71 (C3). ESI-MS calcd for [C24H37O3]−, 373.27; observed, 373.37
3-Oxo-chol-4-ene-24-oic acid (6)
A stock solution of acid 5 (7.5 mg, 20 μmol) was prepared in 2 mL of EtOH. Oxidation of 5 (0.5 mM) was performed in 50 mM sodium phosphate (10 mL, pH 7.5) with 1 μM cholesterol oxidase26 at 30 °C for 14 h. The reaction mixture was extracted with EtOAc (20 mL × 3). The EtOAc extracts were combined, dried over Na2SO4, and concentrated in vacuo to yield 3-oxo-chol-4-ene-24-oic acid (6) (7.37 mg 98%). 1H NMR (500 MHz, CD3OD) δ 5.73 (s, 1H), 1.26 (s, 3H), 0.99 (d, J = 6.6 Hz, 3H), 0.80 (s, 3H). 13C NMR (500 MHz, CD3OD) δ 201.0 (C3), 174.0 (C24), 122.7 (C4). ESI-MS calcd for [C24H35O3]−, 371.26; observed, 371.35.
3-Oxo-chol-4-ene-24-oyl-CoA, 3-OCO-CoA
A stock solution of acid 6 (30 mM) was prepared in EtOH. Thioesterification of 6 (0.66 mM) was performed in 100 mM HEPES buffer (10 mL, pH 8.0) with 2.5 mM ATP, 1.1 mM CoA, 10 mM MgCl2, and 0.5 μM mtFadD1727 at 30 °C for 1 h. The reaction was quenched upon addition of acetic acid to a final concentration of 2% (v/v). The unreacted steroid acid was extracted with EtOAc, and the aqueous layer containing 3-OCO-CoA was directly loaded onto a C18 reverse-phase column (5.5 g C18 gold column, Teledyne ISCO, Ohio), and a linear gradient from 0 to 100% B over 120 mL was run to purify 3-OCO-CoA. A: 0.1 M NH4+OAc–, pH 4.5. B: 9:1 MeOH/1 M NH4+OAc–, pH 4.5. CoA ester 3-OCO-CoA eluted at 100% solvent B. The final product was analyzed by LC/UV/MS, and the purity was greater than 95%. ESI-MS calcd for [C45H69N7O18P3S]−, 1120.36; observed, 1120.38. ESI-MS calcd for [C45H68N7O18P3S]2–, 559.68; observed, 559.75.
(E)-6β-Methoxy-3α,5-cyclo-5α-chol-22-ene-24-aldehyde25 (7)
To a solution of aldehyde 2(24) (70 mg, 0.2 mmol) and ZnBr2 (40 mg, 0.18 mmol) in 3 mL of THF was added N-(t-butyl)-2,2-bis(trimethylsilyl)acetaldimine (50 uL, 1 equiv). After being stirred at 25 °C for 12 h, the resultant imine was hydrolyzed by the addition of 1 to 2 mg of ZnCl2 dissolved in 0.5 mL of H2O and 0.7 mL of Et2O. The solution was stirred for 1 h at 25 °C. The THF/Et2O was evaporated, 5 mL of H2O was added to the residue, and the aqueous solution was extracted with EtOAc (10 mL × 3). The EtOAc layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:19) to yield (E)-6β-methoxy-3α,5-cyclo-5α-chol-22-ene-24-aldehyde (7) (20 mg, 27%). 1H NMR (500 MHz, CDCl3) δ 9.47 (d, J = 7.9 Hz, 1H), 6.71 (dd, J = 15.5, 8.7 Hz, 1H), 6.04 (ddd, J = 15.5, 7.9, 0.7 Hz, 1H), 3.32 (s, 3H), 2.77 (t, J = 2.8 Hz, 1H), 1.13 (d, J = 6.6 Hz, 3H), 1.02 (s, 3H), 0.77 (s, 3H). 13C NMR (500 MHz, CDCl3) δ 194.6 (C24), 164.5 (C22), 130.8 (C23), 82.3 (OMe).
(22E,24E)-Ethyl 6β-Methoxy-3α,5-cyclo-5α-cholest-22,24-dien-26-ate (8)
To a cooled (0 °C, on ice) solution of NaH (3 mg, 0.1 mmol) in 1 mL of THF, ethyl diethylphosphonopropionate (29 mg, 0.1 mmol) was added dropwise. After being stirred for 5 min, a solution of aldehyde 7 (20 mg, 0.054 mmol) in 1 mL of THF was added to the reaction. The reaction mixture was allowed to warm to 25 °C and stirred for an additional 12 h. The THF was evaporated, and the product was purified by flash chromatography (EtOAc/hexane1:19) to yield ethyl (22E,24E)-6β-methoxy-3α,5-cyclo-5α-cholest-22,24-dien-26-ate 8 (25 mg, 92%). 1H NMR (700 MHz, CDCl3) δ 7.14–7.09 (d, J = 11.3, 1H), 6.24 (dd, J = 15.0, 11.3 Hz, 1H), 5.91 (dd, J = 15.0, 8.9 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 3.30 (s, 3H), 1.90 (d, J = 1.1 Hz, 3H), 1.27 (t, J = 7.1, 3H), 1.05 (d, J = 6.6, 3H), 1.01 (s, 3H), 0.74 (s, 3H). 13C NMR (700 MHz, CDCl3) δ 168.6 (C27), 149.2 (C24), 138.9 (C22), 124.9 (C23), 123.4 (C25), 82.3 (OMe).
Ethyl 6β-Methoxy-3α,5-cyclo-5α-cholest-26-ate (9)
A stirred solution of diene 8 (25 mg, 0.052 mmol) and 5 wt % Pd/C (10 mg) in 1 mL of THF was bubbled with H2 for 1 h. The solution was filtered through a pad of Celite and concentrated in vacuo to yield ethyl 6β-methoxy-3α,5-cyclo-5α-cholest-26-ate (9) (25 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 4.14 (q, J = 7.1 Hz, 2H), 3.30 (s, 3H), 1.25 (t, J = 7.1, 3H), 1.14–1.12 (m, 3H), 1.01 (s, 3H), 0.90 (d, J = 6.6, 3H), 0.74 (s, 3H). 13C NMR (400 MHz, CDCl3) δ 176.6 (C27), 82.4 (OMe).
3-Hydroxy-cholest-5-en-26-oic acid (10)
Ester 9 (25 mg, 0.052 mmol) was dissolved in 1 M LiOH (1 mL, dioxane/H2O 7:3), and the reaction mixture was stirred at 25 °C for 12 h. The solution was acidified with 1 N HCl to pH 2, and the product was extracted with EtOAc (10 mL × 3). The EtOAc layers were combined, dried over Na2SO4, and concentrated in vacuo. The solid was dissolved in 1 mL of dioxane/H2O (7:3), p-toluenesulfonic acid (5 mg, 0.5 equiv) was added, and the solution was stirred at 70 °C for 2 h. The product was extracted with EtOAc (5 mL × 3). The EtOAc layers were combined, dried over Na2SO4, and concentrated in vacuo. The final product was purified by flash chromatography (EtOAc/hexane 1:4) to yield 3-hydroxyl-cholest-5-en-26-oic acid 10 (21 mg, 97%). 1H NMR (500 MHz, CDCl3) δ 5.34–5.32 (m, 1H), 3.55–3.49 (m, 1H), 1.17–1.15 (m, 3H), 0.99 (s, 3H), 0.90 (d, J = 6.6, 3H), 0.66 (s, 3H). 13C NMR (500 MHz, CDCl3) δ 182.0 (C27), 140.8 (C5), 121.0 (C6), 71 (C3).
3-Oxo-cholest-4-en-26-oic acid (11)
The procedure for the preparation of 6 from 5 described above was followed using acid 10 to prepare 11. 1H NMR (500 MHz, CDCl3) δ 5.72 (s, 1H), 1.19–1.18 (m, 6H), 0.90 (d, J = 6.6, 3H), 0.70 (s, 3H). 13C NMR (500 MHz, CDCl3) δ 199.8 (C3), 171.8 (C27), 123.7 (C4). ESI-MS calcd for [C27H41O3]−, 413.31; observed, 413.34.
3-Oxo-cholest-4-en-26-oyl CoA, 3-OCS-CoA
The procedure for the preparation of 7 from 6 described above was followed using acid 11 to prepare 3-OCS-CoA, except that mtFadD19 was used. mtFadD19 was cloned, expressed, and purified as described for mtFadD17.27 ESI-MS calcd for [C48H75N7O18P3S]−, 1162.41; observed, 1162.42.
Expression Plasmid Construction
Genes chsE4 (Rv3504), chsE5 (Rv3505), and chsE3 (Rv3573c) were amplified from M. tuberculosis H37Rv total genomic DNA by PCR using forward and reverse primers. The PCR product was digested with the appropriate restriction endonuclease and ligated into a similarly digested pET28b or pET20b vector (Table S1). DNA sequencing of the plasmids confirmed that the sequence was correct and that no mutations were introduced during cloning. ChsE4 glutamate 247 was mutated to alanine in p26N/27 using the method of quick change site-directed mutagenesis.23 The mutations were confirmed by DNA sequencing.
chsE4 and chsE5 Gene Coexpression and Protein Purification
Constructs p26N and p27 were cotransformed into the BL21(DE3) E. coli cell for in trans expression. Gene coexpression in cis with construct p26N/27 was performed as reported previously.19 Similarly, the ChsE4E247A-ChsE5 mutant protein was prepared with construct p26NE247A/27 using the same expression conditions that were used for p26N/27. Expression was induced at OD600 ≈ 0.6–0.8 by the addition of 1 mM IPTG, and cells were grown for 20 h at 25 °C. Cells were lysed by cell disruption in buffer A, and cellular debris was removed by ultracentrifugation at 125 000g for 1 h. The ChsE4-ChsE5 complex was purified by IMAC, with a HisTrap FF column using buffers A for binding and B for elution. Protein was further purified by size exclusion chromatography on a Superdex 200 HiLoad 16/60 column equilibrated with buffer C.
Protein Expression and Purification
To express ChsE3, construct p34N was transformed into BL21(DE3) E. coli. Single colonies were selected on LB plates containing 30 μg/mL kanamycin and cultured in 2 × YT media at 37 °C. Protein expression was induced at OD600 ≈ 0.6–0.8 by the addition of 50 μM to 1 mM isopropyl 1-thio-β-d-galactopyranoside (IPTG), and cells were grown for 20 h at 25 °C. Purified proteins were analyzed by reducing SDS-PAGE. The protein was purified using a method similar to that described for ChsE4-ChsE5 purification.
Selenomethionine-Substituted ChsE4 and ChsE5 Coexpression and Protein Purification
Construct p26N/27 was transformed into BL21 (DE3) E. coli cell for in cis expression. Single colonies were selected on LB plates containing 30 μg/mL kanamycin and cultured in M9 minimal media supplemented with 30 μg/mL kanamycin. Selenomethionine (SeMet) and other essential amino acids were added when the OD600 value reached ∼1.1, and then cells were grown at 18 °C for 20 h. SeMet-substituted ChsE4-ChsE5 protein was purified in the same manner as was the native protein.
Solution-State Biophysical Analysis of ChsE3 and ChsE4-ChsE5
Molecular weights were determined using analytical ultracentrifugation sedimentation equilibrium (Beckman Optima XL-A). ChsE4-ChsE5 (5.18, 2.28, and 1.08 μM) and ChsE3 (6.09, 3.05, and 1.52 μM) were centrifuged at speeds of 20K, 25K, and 30K at 20 °C. Scans were acquired after 18 and 20 h of centrifugation at each speed, with monitoring at 280 nm. Approximately 80 curves generated from replicate scans were fit globally in Heteroanalysis (University of Connecticut, Storrs, CT) to the ideal single-species model. The sample buffer density (ρ) and partial specific volume (ν) of each protein were calculated using SEDNTERP (University of New Hampshire, Durham, NH).
Protein complex stoichiometries of ChsE4-ChsE5 were confirmed by LC/UV/MS. Samples were separated on a XBridge BEH 300 C4 3.5 μm column (2.1 × 100 mm) at 40 °C with a linear gradient from 95% A to 95% B over 15 min, where A is 5% isopropanol/0.1% trifluoroacetic acid and B is 99.9% isopropanol/0.1% trifluoroacetic acid. MS spectra were collected in ESI positive ion mode with a cone voltage of 40 V, a capillary voltage of 4.5 kV, and a source temperature of 150 °C. MS spectra were deconvoluted using ESIprot 1.0,37 and peaks in the UV (280 nm) were integrated using R. The integrated peak areas of each protein were divided by the corresponding molar extinction coefficient for the protein to yield the molar concentrations. Protein stoichiometries were determined from the ratio of the molar concentrations.
Determination of the Protein to FAD Ratio
The oligomeric states of the ChsE4-ChsE5 complex and the ChsE3 to FAD ratio were determined using the method described previously.19
Dehydrogenase Assay
The dehydrogenase activities of ChsE1-ChsE2, ChsE3, and ChsE4-ChsE5 (50 mM) were tested individually with 3-OPC-CoA, 3-OCO-CoA, and 3-OCS-CoA (1–100 μM) with artificial electron acceptor ferricenium hexafluorophosphate (250 μM, extinction coefficient 3.4 mM–1 cm–1)28 in 100 mM TAPS buffer (pH 8.5) as reported previously.16,17 Assays were initiated by the addition of enzyme. Product formation was monitored spectroscopically at 300 nm at 25 °C. Initial velocities were obtained for the first 10–15% of the reaction. Controls were run without enzyme or without substrate and showed negligible decreases in absorbance at 300 nm. The rates of product formation were fit to the Michaelis–Menten equation to determine KM and kcat for each substrate, except in the case of ChsE4-ChsE5/3-OCS-CoA, for which diastereomer inhibition was observed at high concentrations. In that case, data were fit to uncompetitive inhibition eq 1 for which [S] = [I] = the concentration of a single diastereomer:
| 1 |
Protein Crystallization
The ChsE4-ChsE5 apoenzyme crystals were obtained by hanging drop vapor diffusion at room temperature. Briefly, 1 μL of a 12 mg mL–1 protein was mixed 1:1 with a reservoir solution of 25% v/v poly(ethylene glycol) monomethyl ether 550, 0.005 M MgCl2·6H2O, and 0.05 M HEPES sodium pH 7.0 and equilibrated against 500 μL of the reservoir solution. The crystals were then harvested and transferred to a cryoprotectant solution containing 27% v/v poly(ethylene glycol) monomethyl ether 550, 0.005 M MgCl2·6H2O, 0.05 M HEPES sodium pH 7.0, and 10% glycerol. All crystals were cryocooled in liquid N2 before data collection.
X-ray Data Collection and Structural Determination
Diffraction data were collected on Beamline X25 of the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY) at the selenium absorption edge using a wavelength of 0.9792 Å. Data sets were processed using XDS38 and Aimless39 as implemented in the autoPROC pipeline.40 All 26 selenium sites were located with SHELXD.41 Phases were calculated to 2.0 Å via a single-wavelength anomalous dispersion42 using Sharp.43 An initial round of density modification and automated model building was carried out with Solomon,44 Buccaneer,45 and Parrot.46 The resulting map and model were further improved in a second round of density modification and automated model building in Solomon and ARP/wARP. This procedure yielded a starting model that was 94% complete (1408 of 1496 residues correctly built). Manual model adjustments were carried out in Coot47 followed by refinement with Refmac48 and Phenix.49 The geometric quality of the refined model was assessed with MolProbity50 and the structure validation tools in the Phenix suite. Data collection and refinement statistics are shown in Table 2.
Single-Crystal Optical Spectra Collection during the Course of X-ray Diffraction Data Collection
Single-crystal optical spectra for ChsE4-ChsE5 crystals were collected on beamline X26-C of the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY) using X-ray exposure at a wavelength of 1.0 Å. With 72 different optical spectra taken every 5° by rotating the crystal 360°, the optimal spectroscopic angle for the cryoloop and crystal was defined using CBASS software.51 X-ray diffraction collection started with 180 × 1 °C rotation to acquire a full data set. During the readout of X-ray diffraction images, correlated electronic absorption spectra were collected with the crystal rotating to the optimal angle. A set of optical spectra were overlaid and compared to the cumulative X-ray exposure time after full data collection.37
Small-Angle X-ray Scattering of ChsE4-ChsE5 in Solution
Purified ChsE4-ChsE5 was concentrated to 72.4 μM (12.3 mg/mL). Small-angle X-ray scattering (SAXS) data sets were collected for both the ChsE4-ChsE5 solutions (72.4, 36.2, and 18.1 μM) and the buffer as a reference at the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY) on beamline X9 using an energy of 13.5 keV.52 Solutions were transferred to a 96-well plate and placed in a water-thermostated sample holder (10 °C). Throughout the course of the experiment (about 30 s per run), samples flowed continuously through a 0.9 mm quartz capillary tube to minimize damage from X-ray radiation (∼20 μL).
The scattering contribution of the ChsE4-ChsE5 protein complex was obtained by subtracting the buffer scattering profile from the protein solution scattering profile. Data processing was performed using a Python-based package developed at X9. The data were fit in GNOM to produce a well-behaved P(r) curve. P(r) curves were compared for three different concentrations in PRIMUS. Twenty protein-shaped (bead) models were reconstructed in DAMMIF and then aligned and compared in DAMSEL in order to determine the most probable one. DAMSUP was used to align all models with the most probable one, and all of the aligned models were averaged in DAMAVER to compute a probability map. DAMFILT was applied to filter the average model at a preset cutoff volume. The DAMFILT models were used to yield the final SAXS envelope structures.
Docking of 3-Oxo-cholest-4-en-26-oyl-CoA into the Crystal Structure of ChsE4-ChsE5
The crystal structure of a medium chain acyl-CoA dehydrogenase (MCAD) with bound octanoyl-CoA (PDB code: 3MDE) was superimposed onto the apo-ChsE4-ChsE5 structure in PyMOL (PyMOLX11Hybrid) using a backbone alignment. Octanoyl-CoA was extracted and merged with the apo-ChsE4-ChsE5 structure. The new complex was further analyzed in Chimera (UCSF Chimera) using default parameters of Dock Prep tool to dock ligand (25S)-3-OCS-CoA into the ChsE4-ChsE5 structure. To add charge to the ligand, AMBER ff99SB and gasteiger were used as the charge model and charge method, respectively. SPHGEN was used to generate receptor spheres. During docking, the conserved position of coenzyme A was fixed and used as a constraint. Dock6 was used to finish the docking. Consensus scoring and manual inspection were used to select docking poses for further analysis.
Acknowledgments
This work was supported by NIH AI092455 (N.S.S.), NIH AI085349 (N.S.S.), NIH RR021008 (N.S.S.), NIH ES022930 (K.E.G), NIH GM100021 (M.G-D.), and NSF BIO1039771. Use of beamlines x9, x25, and x26 at the National Synchrotron Light Source of Brookhaven National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-98CH10886. Use of the GM/CA beamlines (23ID-B and 23ID-D) at the Advanced Photon Source, Argonne National Laboratory was funded in whole or in part with federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of the Advanced Photon Source facilities was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science under contract no. DE-AC02-06CH11357. We thank all members of the Sampson and Garcia-Diaz laboratory for insightful discussion and support and Nukri Sanishvili and the CCP4 school at APS for assistance with X-ray data collection.
Supporting Information Available
The following file is available free of charge on the ACS Publications website at DOI: 10.1021/id500033m.
Six additional figures, one table, and 1H and 13C spectra of synthetic intermediates and products (PDF)
Accession Codes
Atomic coordinates and structure factors for ChsE4-ChsE5/FADH2 have been deposited in the Protein Data Bank with accession code 4X28.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nesbitt N. M.; Yang X.; Fontán P.; Kolesnikova I.; Smith I.; Sampson N. S.; Dubnau E. (2010) A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect. Immun. 78, 275–282 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam K. C.; D’Angelo I.; Kalscheuer R.; Zhu H.; Wang J.-X.; Snieckus V.; Ly L. H.; Converse P. J.; Jacobs W. R.; Strynadka N.; Eltis L. D. (2009) Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 5, e1000344. 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A. K.; Sassetti C. M. (2008) Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U.S.A. 105, 4376–4380 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. C.; Miner M. D.; Pandey A. K.; Gill W. P.; Harik N. S.; Sassetti C. M.; Sherman D. R. (2009) igr Genes and Mycobacterium tuberculosis Cholesterol Metabolism. J. Bact. 191, 5232–5239 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron P.; Vaubourgeix J.; Poquet Y.; Levillain F.; Botanch C.; Bardou F.; Daffe M.; Emile J. F.; Marchou B.; Cardona P. J.; de Chastellier C.; Altare F. (2008) Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 4, e1000204. 10.1371/journal.ppat.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sih C. J.; Tai H. H.; Tsong Y. Y. (1967) The mechanism of microbial conversion of cholesterol into 17-keto steroids. J. Am. Chem. Soc. 89, 1957. 10.1021/ja00984a039. [DOI] [PubMed] [Google Scholar]
- Sih C. J.; Tai H. H.; Tsong Y. Y.; Lee S. S.; Coombe R. G. (1968) Mechanisms of steroid oxidation by microorganisms 0.14. Pathway of cholesterol side-chain degradation. Biochemistry 7, 808–818 10.1021/bi00842a039. [DOI] [PubMed] [Google Scholar]
- Reddy J. K.; Hashimoto T. (2001) Peroxisomal β-oxidation and peroxisome proliferator-activated receptor α: An Adaptive Metabolic System. Annu. Rev. Nutrit. 21, 193–230 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- Yang X.; Dubnau E.; Smith I.; Sampson N. S. (2007) Rv1106c from Mycobacterium tuberculosis Is a 3β-Hydroxysteroid Dehydrogenase. Biochemistry 46, 9058–9067 10.1021/bi700688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capyk J. K.; Kalscheuer R.; Stewart G. R.; Liu J.; Kwon H.; Zhao R.; Okamoto S.; Jacobs W. R.; Eltis L. D.; Mohn W. W. (2009) Mycobacterial Cytochrome P450 125 (Cyp125) Catalyzes the Terminal Hydroxylation of C27 Steroids. J. Biol. Chem. 284, 35534–35542 10.1074/jbc.M109.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. B.; Ouellet H.; Ortiz de Montellano P. R. (2010) Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J. Biol. Chem. 285, 36352–36360 10.1074/jbc.M110.161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet H.; Guan S.; Johnston J. B.; Chow E. D.; Kells P. M.; Burlingame A. L.; Cox J. S.; Podust L. M.; De Montellano P. R. O. (2010) Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol. Microbiol. 77, 730–742 10.1111/j.1365-2958.2010.07243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wilbrink M. H.; Petrusma M.; Dijkhuizen L.; van der Geize R. (2011) FadD19 of Rhodococcus rhodochrous DSM43269, a steroid-coenzyme A ligase essential for degradation of C-24 branched sterol side chains. Appl. Environ. Microbiol. 77, 4455–4464 10.1128/AEM.00380-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Casabon I.; Swain K.; Crowe A. M.; Eltis L. D. (2014) Actinobacterial acyl coenzyme A synthetases involved in steroid side-chain catabolism. J. Bacteriol. 196, 579–587 10.1128/JB.01012-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Geize R.; Yam K.; Heuser T.; Wilbrink M. H.; Hara H.; Anderton M. C.; Sim E.; Dijkhuizen L.; Davies J. E.; Mohn W. W.; Eltis L. D. (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. U.S.A. 104, 1947–1952 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisla S.; Thorpe C. (2004) Acyl-CoA dehydrogenases. A mechanistic overview. Eur. J. Biochem. 271, 494–508 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- Thomas S. T.; VanderVen B. C.; Sherman D. R.; Russell D. G.; Sampson N. S. (2011) Pathway Profiling in Mycobacterium tuberculosis: Elucidation of a cholesterol-derived catabolite and the enzymes that catalyze its metabolism. J. Biol. Chem. 286, 43668–43678 10.1074/jbc.M111.313643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. T.; Sampson N. S. (2013) Mycobacterium tuberculosis utilizes a unique heterotetrameric structure for dehydrogenation of the cholesterol side chain. Biochemistry 52, 2895–2904 10.1021/bi4002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Guja K. E.; Thomas S. T.; Garcia-Diaz M.; Sampson N. S. (2014) A Distinct MaoC-like enoyl-CoA hydratase architecture mediates cholesterol catabolism in Mycobacterium tuberculosis. ACS Chem. Biol. 10.1021/cb500232h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipperman M. F.; Yang M.; Thomas S. T.; Sampson N. S. (2013) Shrinking the FadE proteome of Mycobacterium tuberculosis: Insights into cholesterol metabolism through identification of an α2β2 heterotetrameric acyl coenzyme A dehydrogenase family. J. Bact. 195, 4331–4341 10.1128/JB.00502-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall S. L.; Withers M.; Soffair C. N.; Moreland N. J.; Gurcha S.; Sidders B.; Frita R.; Ten Bokum A.; Besra G. S.; Lott J. S.; Stoker N. G. (2007) A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol. Microbiol. 65, 684–699 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Ge F.; Zhang Q.; Ren Y.; Yuan J.; He J.; Li W.; Chen G.; Zhang G.; Zhuang Y.; Xu L. (2014) Identification of gene expression profiles in the actinomycete Gordonia neofelifaecis grown with different steroids. Genome 57, 345–353 10.1139/gen-2014-0030. [DOI] [PubMed] [Google Scholar]
- Kendall S. L.; Burgess P.; Balhana R.; Withers M.; Ten Bokum A.; Lott J. S.; Gao C.; Uhia-Castro I.; Stoker N. G. (2010) Cholesterol utilization in mycobacteria is controlled by two TetR-type transcriptional regulators: kstR and kstR2. Microbiology 156, 1362–1371 10.1099/mic.0.034538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-J. P.; Miura R. (2004) Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. Eur. J. Biochem. 271, 483–493 10.1046/j.1432-1033.2003.03948.x. [DOI] [PubMed] [Google Scholar]
- Salmond W. G.; Sobala M. C. (1977) An efficient synthesis of 25-hydroxycholesterol from stigmasterol. Tetrahedron Lett. 18, 1695–1698 10.1016/S0040-4039(01)93251-4. [DOI] [Google Scholar]
- Bellassoued M.; Majidi A. (1993) A simple and highly stereoselective route to E-α,β-unsaturated aldehydes. J. Org. Chem. 58, 2517–2522 10.1021/jo00061a027. [DOI] [Google Scholar]
- Yue Q. K.; Kass I. J.; Sampson N. S.; Vrielink A. (1999) Crystal structure determination of cholesterol oxidase from Streptomyces and structural characterization of key active site mutants. Biochemistry 38, 4277–4286 10.1021/bi982497j. [DOI] [PubMed] [Google Scholar]
- Schaefer C.; Lu R.; Nesbitt N. M.; Schiebel J.; Sampson N. S.; Kisker C. (2014) FadA5 a thiolase from Mycobacterium tuberculosis – a unique steroid-binding pocket reveals the potential for drug development against tuberculosis. Structure 10.1016/j.str.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman T. C.; Hale D. E.; Bhala A.; Thorpe C. (1990) An acyl-coenzyme A dehydrogenase assay utilizing the ferricenium ion. Anal. Biochem. 186, 280–284 10.1016/0003-2697(90)90080-S. [DOI] [PubMed] [Google Scholar]
- Schmitz W.; Conzelmann E. (1997) Stereochemistry of peroxisomal and mitochondrial beta-oxidation of alpha-methylacyl-CoAs. Eur. J. Biochem. 244, 434–440 10.1111/j.1432-1033.1997.00434.x. [DOI] [PubMed] [Google Scholar]
- Pascual C.; Meier J.; Simon W. (1966) Regel zur Abschatzung der Chemischen Verschiebung von Protonen an einer Doppelbindung. Helv. Chim. Acta 49, 164–168 10.1002/Hlca.660490122. [DOI] [Google Scholar]
- Karplus P. A.; Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. J.; Wang M.; Paschke R. (1993) Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc. Natl. Acad. Sci. U.S.A. 90, 7523–7527 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlikova B.; Sebestova E.; Sustr V.; Brezovsky J.; Strnad O.; Daniel L.; Bednar D.; Pavelka A.; Manak M.; Bezdeka M.; Benes P.; Kotry M.; Gora A.; Damborsky J.; Sochor J. (2014) CAVER Analyst 1.0: graphic tool for interactive visualization and analysis of tunnels and channels in protein structures. Bioinformatics 30, 2684–2685 10.1093/bioinformatics/btu364. [DOI] [PubMed] [Google Scholar]
- Chang J. C.; Harik N. S.; Liao R. P.; Sherman D. R. (2007) Identification of Mycobacterial genes that alter growth and pathology in macrophages and in mice. J. Infect. Dis. 196, 788–795 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- Bennett B. D.; Kimball E. H.; Gao M.; Osterhout R.; Van Dien S. J.; Rabinowitz J. D. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipperman M. F.; Sampson N. S.; Thomas S. T. (2014) Pathogen roid rage: Cholesterol utilization by Mycobacterium tuberculosis. Crit. Rev. Biochem. Mol. Biol. 49, 269–293 10.3109/10409238.2014.895700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orville A. M.; Buono R.; Cowan M.; Héroux A.; Shea-McCarthy G.; Schneider D. K.; Skinner J. M.; Skinner M. J.; Stoner-Ma D.; Sweet R. M. (2011) Correlated single-crystal electronic absorption spectroscopy and X-ray crystallography at NSLS beamline X26-C. J. Synchrotron Rad. 18, 358–366 10.1107/S0909049511006315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., D 66, 125–132 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. (2013) How good are my data and what is the resolution?. Acta Crystallogr., D 69, 1204–1214 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr., D 67, 293–302 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T. R.; Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr., D 58, 1772–1779 10.1107/S0907444902011678. [DOI] [PubMed] [Google Scholar]
- Dauter Z.; Dauter M.; Dodson E. (2002) Jolly SAD. Acta Crystallogr., D 58, 494–506 10.1107/S090744490200118X. [DOI] [PubMed] [Google Scholar]
- Vonrhein C., Blanc E., Roversi P., and Bricogne G. (2007) Automated Structure Solution with autoSHARP, in Macromolecular Crystallography Protocols (Doublié S., Ed.), pp 215–230, Humana Press. [DOI] [PubMed] [Google Scholar]
- Abrahams J. P.; Leslie A. G. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr., D 52, 30–42 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr., D 62, 1002–1011 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Cowtan K. (2010) Recent developments in classical density modification. Acta Crystallogr., D 66, 470–478 10.1107/S090744490903947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., D 66, 486–501 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., D 67, 355–367 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart P., Afonine P., Grosse-Kunstleve R., Hung L.-W., Ioerger T., McCoy A., McKee E., Moriarty N., Read R., Sacchettini J., Sauter N., Storoni L., Terwilliger T., and Adams P. (2008) Automated Structure Solution with the PHENIX Suite, in Structural Proteomics (Kobe B., Guss M., and Huber T., Eds.), pp 419–435, Humana Press: DOI: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. III; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., D 66, 12–21 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner J. M.; Cowan M.; Buono R.; Nolan W.; Bosshard H.; Robinson H. H.; Heroux A.; Soares A. S.; Schneider D. K.; Sweet R. M. (2006) Integrated software for macromolecular crystallography synchrotron beamlines II: revision, robots and a database. Acta Crystallogr., D 62, 1340–1347 10.1107/S0907444906030162. [DOI] [PubMed] [Google Scholar]
- Allaire M.; Yang L. (2011) Biomolecular solution X-ray scattering at the National Synchrotron Light Source. J. Synchrotron Radiat. 18, 41–44 10.1107/S0909049510036022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.