

Abstract
Background
Small heterodimer partner (SHP, NR0B2) is involved in diverse metabolic pathways, including hepatic bile acid, lipid and glucose homeostasis, and has been implicated in effects on the peroxisome proliferator-activated receptor γ (PPARγ), a master regulator of adipogenesis and the receptor for antidiabetic drugs thiazolidinediones (TZDs). In this study, we aim to investigate the role of SHP in TZD response by comparing TZD-treated leptin-deficient (ob/ob) and leptin-, SHP-deficient (ob/ob;Shp−/−) double mutant mice.
Results
Both ob/ob and double mutant ob/ob;Shp−/− mice developed hyperglycemia, insulin resistance, and hyperlipidemia, but hepatic fat accumulation was decreased in the double mutant ob/ob;Shp−/− mice. PPARγ2 mRNA levels were markedly lower in ob/ob;Shp−/− liver and decreased to a lesser extent in adipose tissue. The TZD troglitazone did not reduce glucose or circulating triglyceride levels in ob/ob;Shp−/− mice. Expression of the adipocytokines, such as adiponectin and resistin, was not stimulated by troglitazone treatment. Expression of hepatic lipogenic genes was also reduced in ob/ob;Shp−/− mice. Moreover, overexpression of SHP by adenovirus infection increased PPARγ2 mRNA levels in mouse primary hepatocytes.
Conclusions
Our results suggest that SHP is required for both antidiabetic and hypolipidemic effects of TZDs in ob/ob mice through regulation of PPARγ expression.
Keywords: SHP, Troglitazone, PPARγ, Thiazolidinedione
Background
Obesity is associated with cardiovascular disease, type 2 diabetes mellitus and some cancers [1,2]. Among these, type 2 diabetes mellitus is a major source of mortality in the obese population. Diabetes develops in the context of both insulin resistance and β- cell dysfunction [3]. In insulin resistance, the ability of insulin to enhance glucose disposal in muscle and adipose tissue and to decrease gluconeogenesis in liver is impaired. Diabetes ensues when the pancreatic β-cell cannot compensate for insulin resistance by adequately increasing insulin secretion.
Thiazolidinediones (TZDs) are a class of antidiabetic drugs that act by increasing insulin sensitivity [4,5]. TZDs, including troglitazone, rosiglitazone and pioglitazone, improve insulin action in patients and a number of insulin-resistant obese and diabetic murine models, such as ob/ob (leptin-deficient), db/db (leptin receptor-deficient), KKAy mice and Zucker fatty rats [6-8]. TZDs are potent agonist ligands for the nuclear hormone receptor peroxisome proliferator-activated receptor γ (PPARγ) and their antidiabetic actions are believed to be mediated by interactions with PPARγ [9,10]. PPARγ is a key regulator of adipogenesis [11,12] that exists as two protein isoforms, PPARγ1 and γ2, arising from differential promoter usage. PPARγ2 encodes 30 additional amino acids at the N-terminus compared to PPARγ1. PPARγ2 is expressed at highest levels in adipose tissue compared to other major insulin target tissues, such as liver and muscle, whereas PPARγ1 is expressed at relatively low levels in many tissues [11,13]. The expression pattern suggests that adipose tissue is the primary target of TZD-induced insulin sensitization as generally supported by tissue-specific PPARγ knockout studies, although other tissues and cell types also contribute [14-18]. PPARγ expression levels can change under different physiological conditions and affect the response to TZD treatment [13]. For example, hepatic PPARγ expression is elevated in animals that develop fatty livers [18-20], and increased PPARγ2 expression is correlated with increased liver fat in human subjects with non-alcoholic fatty liver disease (NAFLD) [21]. TZD effects could be amplified in such PPARγ-rich fatty livers, which may be particularly relevant for the beneficial effects of TZD treatment in human patients with non-alcoholic steatohepatitis (NASH) [22,23].
Mutations in the small heterodimer partner (SHP, NR0B2) have been associated with mild obesity in several human populations [24-27]. SHP is an atypical orphan nuclear receptor that lacks a conventional DNA-binding domain [28,29]. Although SHP interacts with several nuclear receptors by acting as a repressor [28,30-32], it has been reported to increase the transcriptional activation of PPARγ [33]. In addition, hepatic PPARγ gene expression is upregulated in transgenic mice expressing SHP in the liver, suggesting that SHP may affect PPARγ expression at the transcription level [34].
To investigate the role of SHP in TZD response in obese diabetic mice, we compared glucose metabolism and lipid profiles in ob/ob and ob/ob;Shp−/− double mutant mice after TZD treatment. Troglitazone did not reduce glucose or circulating triglyceride levels in the ob/ob;Shp−/− mice, which showed markedly decreased PPARγ expression in liver and, to a lesser extent, adipose tissue. Furthermore, SHP overexpression increased PPARγ mRNA levels in primary hepatocytes. These results suggest that SHP is required for TZD effects in ob/ob mice and for a potential indirect activation of PPARγ gene.
Methods
Animals and treatments
Shp−/− mice were generated previously in this laboratory with a mixed C57BL/129sv hybrid background [35]. They were backcrossed to C57BL/6 J mice for 10 generations to obtain >99.99% pure C57BL/6 J background. The leptin-deficient ob/ob and ob/ob;Shp−/− mice were generated as described previously [36]. Groups of 10–15 male ob/ob and ob/ob;Shp−/− mice (7- to 8-week-old) were oral gavaged with vehicle (10% dimethylsulfoxide (DMSO) in corn oil) or troglitazone (Cayman chemical, dissolved in DMSO, 10 mg/kg/day) for two weeks. Before the first day of treatment and on the day before sacrifice, mice were fasted overnight and blood samples were collected from the orbital plexus. Livers and white adipose tissue were dissected, weighed and fixed for histological analysis, or snap frozen in liquid nitrogen and stored at −80°C until use. Mice were maintained in the accredited pathogen-free facility at Baylor College of Medicine on a 12-hour light/dark cycle and fed a standard rodent chow and water ad libitum. All protocols for animal use were approved by the animal care committee of Baylor College of Medicine.
Histological analysis
Livers were fixed, dehydrated and embedded in paraffin. Sections were cut with a thickness of 5 μm and stained with Harris hematoxylin-eosin (Sigma).
Serum and tissue chemistry
Serum was prepared from whole blood and stored at −80°C until use. Lipids were extracted from liver using chloroform-methanol extraction [37]. Enzymatic assay kits were used for the determination of non-esterified fatty acids (Wako), cholesterol and triglyceride (Thermo Electron). Insulin levels were measured by a mouse/rat insulin ELISA kit (Millipore-Linco).
Glucose tolerance test
Glucose tolerance tests (GTT) were performed by intraperitoneal injection of glucose (2 g/kg of body weight) following overnight fasting. Blood samples were taken at 0, 15, 30, 60, 120 minutes from the tail vein and were analyzed for glucose concentrations using kits from Thermo Electron.
RNA isolation and real-time quantitative PCR
Total RNA was isolated using TRIzol reagent (Invitrogen). 1 μg of total RNA was reverse transcribed using QuantiTect Reverse Transcription kit (Qiagen) according to manufacturer’s instructions. Real-time quantitative PCR (SYBR green) analysis was performed on an ABI prism 7700 sequence detection system (Applied Biosystems) under factory default thermal cycling conditions (50°C, 2 min; 95°C, 10 min; and 40 cycles at 95°C, 15 s; 60°C, 1 min). Expression was normalized to 36B4 and the relative quantification was calculated using ΔΔCt formula.
Primary hepatocyte isolation, culture and adenoviral transduction
Primary hepatocytes were prepared from 8- to 12-week-old wild type mice by in situ perfusion and single-step Percoll gradient centrifugation [35]. Cells were plated at 106 per 6-cm dish and grown in Williams’ E medium supplemented with 10 μg/ml transferrin, 10 μg/ml insulin, 100U/ml penicillin and 100 μg/ml streptomycin. One day after plating, the cells were infected with a SHP-expressing adenovirus or a control virus expressing GFP as described [38] for two hours at a multiplicity of infection (MOI) of 20. Virus-containing media were removed and cells were cultured for two days after infection. Total RNA were isolated from cells for real-time quantitative PCR analysis.
Statistical analysis
Values are presented as means ± SEM. Statistical significance was determined by two-tailed t test or ANOVA, as appropriate.
Results
Troglitazone does not improve the diabetic syndromes in ob/ob;Shp−/− mice
The ob/ob mouse is a valuable type 2 diabetes model. Based on the role of the orphan nuclear receptor SHP in metabolic pathways, we generated ob/ob;Shp−/− double mutant mice. The obesity of the double mutants was not different from the ob/ob mice (9-10-week-old ob/ob;Shp−/− body weight 38.38 ± 1.9 g versus ob/ob 35.8 ± 1.7 g). We initially assessed the effects of SHP deficiency on glucose homeostasis by measuring blood glucose and insulin levels. Glucose levels of ob/ob;Shp−/− mice were significantly higher than those of ob/ob mice, whereas the insulin level was markedly lower (Figure 1A, B). To further characterize glucose metabolism, glucose tolerance tests were performed and ob/ob;Shp−/− mice were more glucose-intolerant compared to ob/ob mice (Figure 1C). These results suggest that SHP deficiency aggravates hyperglycemia and insulin resistance in ob/ob mice, which is quite different from the improvements previously described [36]. The basis for this marked discrepancy is not clear.
Figure 1.
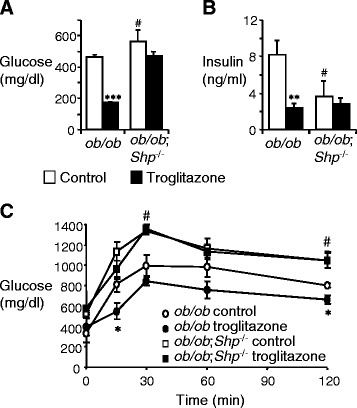
SHP deficiency causes non-responsiveness to antidiabetic effect of TZDs. (A, B) Serum glucose (A) and insulin (B) levels under fasting conditions. 7–8 week-old male ob/ob and ob/ob;Shp −/− mice were treated with control (open bars) or troglitazone (filled bars) for 2 weeks. (C) Glucose tolerance tests. Intraperitoneal glucose tolerance tests were performed on ob/ob and ob/ob;Shp −/− mice treated with control (open symbols) or troglitazone (filled symbols) for 2 weeks. n = 4–5 per group. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 for differences between control and troglitazone-treated ob/ob mice. #P < 0.01 for differences between ob/ob;Shp −/− (with no effect of troglitazone treatment) and control-treated ob/ob mice.
To test whether TZDs are effective in ob/ob;Shp−/− mice with severe glucose intolerance, both ob/ob and ob/ob;Shp−/− mice were treated with troglitazone for 2 weeks. As expected, the ob/ob mice showed dramatically lower serum glucose and insulin levels, as well as improved glucose tolerance (Figure 1). In contrast, neither the serum glucose and insulin levels nor the glucose tolerance was improved in the ob/ob;Shp−/− mice. These results indicate that SHP is required for antidiabetic effects of TZDs in ob/ob mice.
Troglitazone has no effect on the lipid profile of ob/ob;Shp−/− mice
Since SHP is normally highly expressed in the liver, we further investigated the potential effects of SHP deficiency in the ob/ob fatty liver. While the body weight showed no significant difference between ob/ob and ob/ob;Shp−/− mice, the liver weight of ob/ob;Shp−/− mice was significantly lower than that of ob/ob mice, resulting in a smaller liver/body weight ratio (9-10-week-old ob/ob;Shp−/− liver weight 1.67 ± 0.17 g versus ob/ob 2.12 ± 0.18 g, P < 0.05) (Figure 2B). Histological analysis of the liver showed that lipid droplets were much smaller and less numerous in ob/ob;Shp−/− mice than that in ob/ob mice, indicating an improvement of fatty liver in ob/ob;Shp−/− mice (Figure 2A), and this was confirmed by measuring hepatic triglycerides (Figure 2C). These results are consistent with those described previously [36]. Troglitazone treatment of ob/ob mice caused a significant increase in liver/body weight ratio and hepatic triglyceride content (Figure 2B and C). Histological results also revealed that the size and number of lipid droplets were increased by troglitazone treatment in ob/ob mice (Figure 2A). However, these effects of troglitazone were not observed in ob/ob;Shp−/− mice. In addition, the serum triglyceride- and FFA-lowering actions of troglitazone observed in the ob/ob mice were absent in ob/ob;Shp−/− mice (Figure 2D and E). These results confirm that SHP is involved in the development of fatty liver in ob/ob mice and is required for hypolipidemic effects of TZDs.
Figure 2.
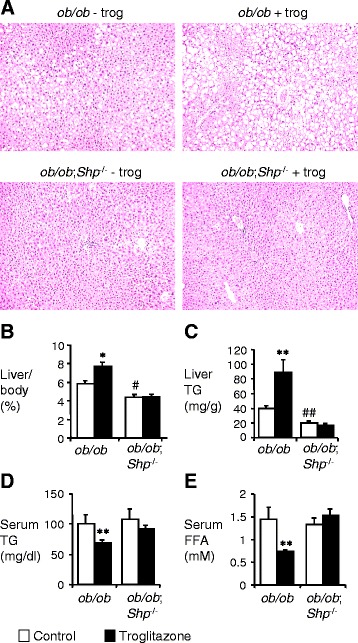
SHP deficiency blunts TZD effects on lipid profile of ob/ob mice. (A) Histology of livers from ob/ob and ob/ob;Shp −/− mice treated with control or troglitazone for 2 weeks. H&E staining was performed for liver sections. ×100 magnification. (B-E) 7–8 week-old male ob/ob and ob/ob;Shp −/− mice were treated with control (open bars) or troglitazone (filled bars) for 2 weeks. (B) Ratio of liver weight to body weight (n = 10–15 per group). (C) Liver triglyceride (TG) content was determined in liver extracts of ob/ob and ob/ob;Shp −/− mice (n = 4–5 per group). (D, E) Serum TG and free fatty acid (FFAs) contents under fasting conditions (n = 10–12 and n = 4–5 per group, respectively). Data are mean ± SEM. *P < 0.01, **P < 0.05, for differences within each genotype between control and troglitazone-treated mice. #P < 0.01, ##P < 0.05 for differences between control-treated ob/ob;Shp −/− and control-treated ob/ob mice.
SHP deficiency downregulates the expression of lipogenic genes in ob/ob mice liver
Since hepatic PPARγ has been reported to play a critical role in the development of fatty liver of ob/ob mice [18], PPARγ1 and γ2 expression was examined in wt, Shp−/−, ob/ob and ob/ob;Shp−/− mice (Figure 3A). The low basal PPARγ1 levels showed about a 3-fold increase in both ob/ob and ob/ob;Shp−/− mice compared to wt mice, but PPARγ2 levels exhibited dramatic differences between ob/ob and ob/ob;Shp−/− mice: a 40-fold increase in ob/ob mice relative to wild type, but only a 5-fold increase in ob/ob;Shp−/− mice.
Figure 3.
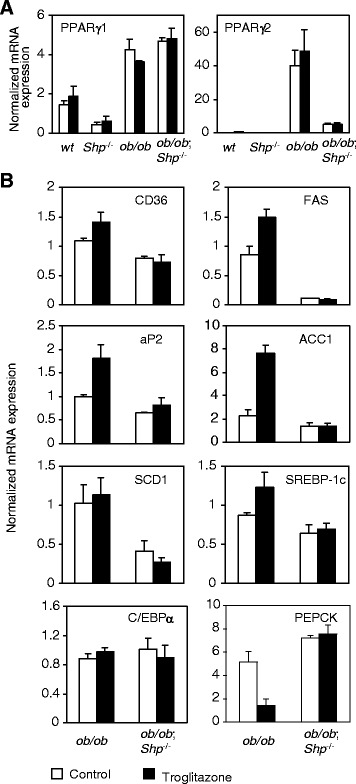
Expression of lipogenic genes was downregulated by SHP deficiency in ob/ob mice liver. Results in panels A and B are liver mRNA levels for control (open bars) and troglitazone-treated (filled bars) mice after 2 weeks of treatment. Data are expressed as relative fold change after normalized to 36B4 and are mean ± SEM (n = 4–5 per group). By two-way ANOVA, the genotype effect (ob/ob and ob/ob;Shp −/−) is significant (P < 0.05) for all except PPARγ1 in panel A. The treatment effect and genotype × treatment interaction is significant for CD36, aP2, FAS, ACC1 and PEPCK in panel B.
To further define the genes regulated by SHP in the ob/ob liver and the mechanism of the decrease in hepatic triglyceride in ob/ob;Shp−/− mice, mRNA from livers of control- and troglitazone-treated mice was analyzed (Figure 3B). mRNA levels of fatty acid translocase (CD36), fatty acid synthase (FAS), adipocyte fatty acid-binding protein (aP2), acetyl-CoA carboxylase 1 (ACC1) and stearoyl-CoA desaturase-1 (SCD-1) were lower in ob/ob;Shp−/− mice than in ob/ob mice. Troglitazone treatment induced the expression of CD36, FAS, aP2 and ACC1 mRNA in ob/ob mice, but not in ob/ob;Shp−/− mice. There was no difference between ob/ob and ob/ob;Shp−/− mice for expression of transcription factors regulating the lipogenic genes, such as SREBP-1c and C/EBPα . The mRNA levels of genes associated with glucose homeostasis, such as phosphoenolpyruvate careboxykinase (PEPCK) for gluconeogenesis, were increased in ob/ob;Shp−/− mice (Figure 3B), which may partly account for the high blood glucose levels in these double mutant mice (Figure 1A). The action of troglitazone to lower glucose levels in type 2 diabetics by decreasing gluconeogenesis in liver was observed in ob/ob mice, but not in ob/ob;Shp−/− mice.
In summary, expression of lipogenic genes was decreased by SHP deficiency in ob/ob mice, which has also been observed in Western diet fed Shp−/− mice [39]. Consistent with the low expression of hepatic PPARγ2 in ob/ob;Shp−/− mice, functional response to PPARγ agonist, troglitazone, was impaired in ob/ob;Shp−/− mice both in lipogenesis and gluconeogenesis.
SHP deficiency affects TZD-responsive gene expression in adipose tissue of ob/ob mice
White adipose tissue has been thought to be the major site of TZD actions, as it is the only insulin-responsive tissue with high expression of PPARγ compared to liver and muscle [18]. Therefore, mRNA levels of genes responsive to TZDs in adipose tissue of control- and troglitazone-treated ob/ob and ob/ob;Shp−/− mice were analyzed by real-time quantitative PCR analysis (Figure 4). Adipose tissue from ob/ob;Shp−/− mice showed an approximately 60% reduction in PPARγ2 expression, which was not as dramatic as the nearly 90% reduction in the liver (Figure 3A). CD36 and adiponectin expression was not different between genotypes, whereas resistin decreased 35% in ob/ob;Shp−/− mice. Troglitazone induced the expression of PPARγ2 and CD36 to a lesser extent in ob/ob;Shp−/− mice than that in ob/ob mice, and failed to induce expression of adiponectin and resistin in ob/ob;Shp−/− mice, demonstrating that SHP is required for full troglitazone responsiveness in adipose tissue.
Figure 4.
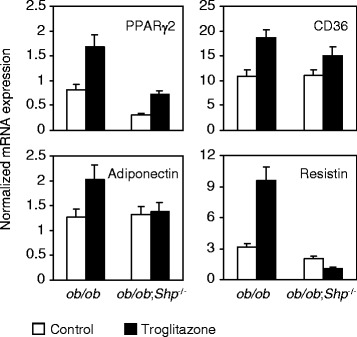
SHP deficiency affects TZD-responsive gene expression in adipose tissue of ob/ob mice. Results are adipose mRNA levels for control (open bars) and troglitazone-treated (filled bars) mice after 2 weeks of treatment. Data are expressed as relative fold change after normalized to 36B4 and are mean ± SEM (n = 4–5 per group). By two-way ANOVA, the genotype and treatment effect is significant (P < 0.05) for PPARγ2 and resistin (treatment effect for CD36, P = 0.06). The genotype × treatment interaction is significant only for resistin. By two-tailed t test, P < 0.05 for differences in adiponectin expression between control and troglitazone-treated ob/ob mice.
SHP upregulates PPARγ2 expression in primary hepatocytes
To test the possibility that SHP might regulate PPARγ2 gene expression, the effects of SHP on the PPARγ2 gene were examined by infecting mouse primary hepatocytes with adenoviral vectors expressing SHP (Figure 5). Transduction of cultured hepatocytes with SHP adenovirus decreased expression of CYP 7A1 mRNA, a known SHP target gene, by 5.2 fold, while increasing PPARγ2 levels for 1.7 fold. These data indicate that SHP overexpression upregulates PPARγ2 expression in primary hepatocytes.
Figure 5.
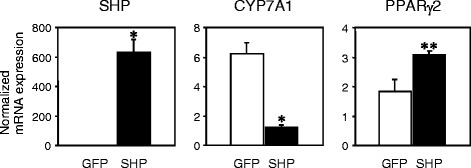
SHP increases PPARγ2 expression in primary hepatocytes. Expression of SHP, CYP7A1 and PPARγ2 in adenovirus-mediated GFP- or SHP-overexpressing hepatocytes by real-time quantitative PCR analysis. Hepatocytes were infected with green fluorescent protein (GFP) or SHP adenovirus as indicated. Data are expressed as relative fold change after normalized to 36B4 and are mean ± SEM (n = 4–5 per group). *P < 0.001, **P < 0.05, compared with GFP-infected cells.
Discussion
The therapeutic use of TZD drugs in the treatment of insulin resistance and type 2 diabetes is now well established [3-5]. TZDs act by increasing insulin sensitivity. These drugs are high affinity ligands for the nuclear receptor PPARγ [9,10] and their antidiabetic effects are thought to be mediated through PPARγ. Therefore, normal PPARγ expression levels, especially in the insulin-responsive tissues, are critical for TZD actions. In this study, we found that SHP deficiency causes downregulation of PPARγ2 expression in liver and adipose tissue of ob/ob mice, and these animals show diminished or abolished responsiveness to the TZD troglitazone. It is reported that mutations in SHP gene in humans are associated with insulin resistance and mild obesity [27]. Our data suggest that the diabetic syndromes of subjects with genetic mutations of SHP may not be improved by TZD treatment. This is the first in vivo evidence that SHP mutation attenuates TZD actions, which makes SHP a possible pharmacogenetic determinant of TZD response.
The ob/ob;Shp−/− double mutant mice showed higher blood glucose levels than ob/ob mice. This may be attributed to abnormal glucose homeostasis in two tissues: skeletal muscle and liver, which have the greatest direct impact on plasma glucose levels. Whereas no difference was observed between ob/ob and ob/ob;Shp−/− mice in gene expression of glucose oxidation and glycogen synthesis in muscle (data not shown), hepatic PEPCK mRNA expression is enhanced in ob/ob;Shp−/− mice compared to ob/ob mice, suggesting that increased gluconeogenesis may contribute to elevated glucose levels. Moreover, the low insulin levels of ob/ob;Shp−/− mice may indicate that pancreatic β cells fail to appropriately compensate for insulin resistance by increasing insulin secretion. It has been shown that independent of PPARγ activation, SHP positively regulates glucose-stimulated insulin secretion (GSIS) in β cells [40], which might be impaired in ob/ob;Shp−/− mice.
The observation that ob/ob;Shp−/− mice exhibit worse hyperglycemia and glucose intolerance than ob/ob mice contrasts with our previous report [36]. In that study, glucose and insulin levels were comparable in both genotypes and loss of SHP was associated with improved insulin sensitivity. One possible explanation is that the age and gender of the mice in the two reports are not exactly the same. The current studies focused solely on male mice at the age of 10 weeks, or older after 2 weeks of control or troglitazone treatment, whereas the prior study used age- and sex-matched groups of younger 8 week old mice. The age of the ob/ob mice may be particularly important since blood glucose rises in this time period before reaching a peak during 3–5 months of age. Thus, it is possible that earlier beneficial effects of the loss of SHP are not evident in these somewhat older mice. Consistent with this, we have observed very similar negative effects of the loss of SHP in long term studies of wild type and Shp−/− mice fed a Western diet [39].
The loss of TZD responsiveness in the ob/ob;Shp−/− mice is presumably a consequence of decreased PPARγ expression. PPARγ1 expression in adipose tissue, liver and muscle is not changed between genotypes, and PPARγ2 expression in muscle is unaffected (Figure 3A and data not shown). Thus, we conclude that loss of SHP primarily affects PPARγ2 expression, which is the dominant isoform in both adipose tissue and fatty liver [41]. Although the effect on PPARγ2 expression was strongest in the liver, a major SHP expressing tissue, decreased hepatic PPARγ2 cannot account for the loss of antidiabetic effects of TZDs, since rosiglitazone improved glucose homeostasis in liver-specific PPARγ knockout mice in the ob/ob background [18]. Although SHP is expressed at only low levels in adipose tissue [33,38], white adipose is the primary target of TZD actions and PPARγ2 expression was significantly decreased in ob/ob;Shp−/− adipose tissue. Consistent with this, the induction of CD36 by troglitazone was decreased, and the response of both adiponectin and resistin was lost in the ob/ob;Shp−/− adipose tissue. Adiponectin promotes fatty acid oxidation and insulin sensitivity in muscle and liver, and the antidiabetic effects of a low dose of pioglitazone were lost in mice in which adiponectin deficiency was introduced into the ob/ob background [42]. Thus, the absence of its induction is likely a major factor in the attenuated effects of troglitazone in the ob/ob;Shp−/− mice. In contrast, elevated resistin has been proposed to increase insulin resistance, and the repression of resistin expression in normal mice by PPARγ agonists is thought to enhance insulin sensitivity [43,44]. Thus, the inductive effect of resistin by troglitazone in the ob/ob mice and the loss of this response in the ob/ob;Shp−/− mice seems inconsistent with the known resistin action. Similar inductive effects of resistin by PPARγ activation have previously been described in both ob/ob mice and Zucker diabetic fatty rats [45], but this was not observed in another study [42]. The basis for these discrepant mRNA expression results, and also a substantial disconnect between resistin adipose mRNA and serum protein levels in ob/ob mice [46], remains unresolved.
SHP is involved in the development of fatty liver by regulating hepatic PPARγ2 and lipogenic genes. The increase in PPARγ2 appears to be a general property of steatotic liver in diet-induced and genetic obese models [18-20,47,48]. Overexpression of PPARγ in a hepatic cell line leads to marked lipid accumulation [49], as does overexpression in PPARα null livers [50]. Thus, SHP deficiency reduced the elevated levels of PPARγ2 and lipogenic genes in ob/ob liver, resulting in the improvement of fatty liver. Additional effects on other pathways may also contribute to the decreased triglyceride accumulation [36].
Recent studies have started to shed light on the molecular basis for the transcriptional regulation of PPARγ by SHP. Renga et al. described that FXR binds to the PPARγ promoter and activates its transcription via the recruitment of SHP in hepatic stellate cells [51]. Whereas Kim et al. identified a novel transcriptional cascade linking RAR/SHP signals with PPARγ2 expression through hairy and enhancer of split 6 (Hes6) in hepatocytes [52]. This transcriptional regulatory pathway controls hepatic lipid metabolism and provides a potential therapeutic entry point for NAFLD. These studies indicate that SHP appears to upregulate PPARγ expression by diverse mechanisms in different cell types.
Conclusions
In summary, our results demonstrate that the antidiabetic and hypolipidemic actions of TZDs require the presence of SHP, likely due to the downregulation of PPARγ2 expression in the adipose tissue and liver of ob/ob;Shp−/− mice. Thus, genetic or pharmacologic modulation of SHP activity could alter the efficacy of TZD antidiabetic actions.
Acknowledgements
We are grateful to J. Liu for assistance with mouse breeding, to Dr. V. K. Yechoor and Dr. B. Chang for assistance in generating adenovirus stocks, and to Drs. J. Lee, K. Ma, M. L. Ricketts, S. A. Johnson and S. Anakk for support and helpful discussions. This work was supported by National Institutes of Health grants R01 DK068804 and U19DK62434 to D.D.M.
Abbreviations
- ACC1
Acetyl-CoA carboxylase
- aP2
Adipocyte fatty acid-binding protein
- CD36
Fatty acid translocase
- C/EBPα
CCAAT/enhancer binding protein alpha
- FAS
Fatty acid synthase
- FXR
Farnesoid X receptor
- GFP
Green fluorescent protein
- FFA
Free fatty acid
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- PEPCK
Phosphoenolpyruvate careboxykinase
- PPARγ
Peroxisome proliferator-activated receptor gamma
- RAR
Retinoic acid receptor
- SCD-1
Stearoyl-CoA desaturase-1
- SHP
Small heterodimer partner
- SREBP-1c
Sterol regulatory element-binding protein 1c
- TG
Triglyceride
- TZDs
Thiazolidinediones
- WT
Wild type
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HTT and DDM conceived and designed experiments, interpreted data and drafted the paper. YJP and YKL participated in the design of the study and contributed to conception. All authors read and approved the final manuscript
Contributor Information
Hsiu-Ting Tseng, Email: httseng@most.gov.tw.
Young Joo Park, Email: yjparkmd@snu.ac.kr.
Yoon Kwang Lee, Email: ylee3@neomed.edu.
David D Moore, Email: moore@bcm.tmc.edu.
References
- 1.Kopelman PG. Obesity as a medical problem. Nature. 2000;404(6778):635–43. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 2.Haslam DW, James WP. Obesity. Lancet. 2005;366(9492):1197–209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 3.Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20(4):573–91. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45(12):1661–9. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- 5.Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest. 2000;106(4):467–72. doi: 10.1172/JCI10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maggs DG, Buchanan TA, Burant CF, Cline G, Gumbiner B, Hsueh WA, et al. Metabolic effects of troglitazone monotherapy in type 2 diabetes mellitus. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1998;128(3):176–85. doi: 10.7326/0003-4819-128-3-199802010-00002. [DOI] [PubMed] [Google Scholar]
- 7.Fujiwara T, Wada M, Fukuda K, Fukami M, Yoshioka S, Yoshioka T, et al. Characterization of CS-045, a new oral antidiabetic agent, II. Effects on glycemic control and pancreatic islet structure at a late stage of the diabetic syndrome in C57BL/KsJ-db/db mice. Metabolism. 1991;40(11):1213–8. doi: 10.1016/0026-0495(91)90218-L. [DOI] [PubMed] [Google Scholar]
- 8.Fujiwara T, Yoshioka S, Yoshioka T, Ushiyama I, Horikoshi H. Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes. 1988;37(11):1549–58. doi: 10.2337/diab.37.11.1549. [DOI] [PubMed] [Google Scholar]
- 9.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270(22):12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 10.Rangwala SM, Lazar MA. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol Sci. 2004;25(6):331–6. doi: 10.1016/j.tips.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid- activated transcription factor. Cell. 1994;79(7):1147–56. doi: 10.1016/0092-8674(94)90006-X. [DOI] [PubMed] [Google Scholar]
- 12.Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, et al. Genetic analysis of adipogenesis through peroxisome proliferator-activated receptor gamma isoforms. J Biol Chem. 2002;277(44):41925–30. doi: 10.1074/jbc.M206950200. [DOI] [PubMed] [Google Scholar]
- 13.Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest. 1997;99(10):2416–22. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Fu M, Cui T, Xiong C, Xu K, Zhong W, et al. Selective disruption of PPARgamma 2 impairs the development of adipose tissue and insulin sensitivity. Proc Natl Acad Sci U S A. 2004;101(29):10703–8. doi: 10.1073/pnas.0403652101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC, et al. Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest. 2003;112(4):608–18. doi: 10.1172/JCI17305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He W, Barak Y, Hevener A, Olson P, Liao D, Le J, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100(26):15712–7. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117(6):1658–69. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111(5):737–47. doi: 10.1172/JCI200317223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao L, Marcus-Samuels B, Mason MM, Moitra J, Vinson C, Arioglu E, et al. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J Clin Invest. 2000;106(10):1221–8. doi: 10.1172/JCI11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, et al. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology. 2000;141(11):4021–31. doi: 10.1210/endo.141.11.7771. [DOI] [PubMed] [Google Scholar]
- 21.Westerbacka J, Kolak M, Kiviluoto T, Arkkila P, Siren J, Hamsten A, et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes. 2007;56(11):2759–65. doi: 10.2337/db07-0156. [DOI] [PubMed] [Google Scholar]
- 22.Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38(4):1008–17. doi: 10.1002/hep.1840380427. [DOI] [PubMed] [Google Scholar]
- 23.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355(22):2297–307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 24.Nishigori H, Tomura H, Tonooka N, Kanamori M, Yamada S, Sho K, et al. Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc Natl Acad Sci U S A. 2001;98(2):575–80. doi: 10.1073/pnas.98.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell SM, Weedon MN, Owen KR, Shields B, Wilkins-Wall B, Walker M, et al. Genetic variation in the small heterodimer partner gene and young-onset type 2 diabetes, obesity, and birth weight in U.K. subjects. Diabetes. 2003;52(5):1276–9. doi: 10.2337/diabetes.52.5.1276. [DOI] [PubMed] [Google Scholar]
- 26.Hung CC, Farooqi IS, Ong K, Luan J, Keogh JM, Pembrey M, et al. Contribution of variants in the small heterodimer partner gene to birthweight, adiposity, and insulin levels: mutational analysis and association studies in multiple populations. Diabetes. 2003;52(5):1288–91. doi: 10.2337/diabetes.52.5.1288. [DOI] [PubMed] [Google Scholar]
- 27.Enya M, Horikawa Y, Kuroda E, Yonemaru K, Tonooka N, Tomura H, et al. Mutations in the small heterodimer partner gene increase morbidity risk in Japanese type 2 diabetes patients. Hum Mutat. 2008;29(11):E271–7. doi: 10.1002/humu.20865. [DOI] [PubMed] [Google Scholar]
- 28.Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272(5266):1336–9. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 29.Seol W, Chung M, Moore DD. Novel receptor interaction and repression domains in the orphan receptor SHP. Mol Cell Biol. 1997;17(12):7126–31. doi: 10.1128/mcb.17.12.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johansson L, Thomsen JS, Damdimopoulos AE, Spyrou G, Gustafsson J, Treuter E. The orphan nuclear receptor SHP inhibits agonist-dependent transcriptional activity of estrogen receptors ERalpha and ERbeta. J Biol Chem. 1999;274(1):345–53. doi: 10.1074/jbc.274.1.345. [DOI] [PubMed] [Google Scholar]
- 31.Lee YK, Dell H, Dowhan DH, Hadzopoulou-Cladaras M, Moore DD. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol Cell Biol. 2000;20(1):187–95. doi: 10.1128/MCB.20.1.187-195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YK, Moore DD. Dual mechanisms for repression of the monomeric orphan receptor liver receptor homologous protein-1 by the orphan small heterodimer partner. J Biol Chem. 2002;277(4):2463–7. doi: 10.1074/jbc.M105161200. [DOI] [PubMed] [Google Scholar]
- 33.Nishizawa H, Yamagata K, Shimomura I, Takahashi M, Kuriyama H, Kishida K, et al. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor gamma transactivation. J Biol Chem. 2002;277(2):1586–92. doi: 10.1074/jbc.M104301200. [DOI] [PubMed] [Google Scholar]
- 34.Boulias K, Katrakili N, Bamberg K, Underhill P, Greenfield A, Talianidis I. Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. Embo J. 2005;24(14):2624–33. doi: 10.1038/sj.emboj.7600728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Lee Y-K, Bundman D, Han Y, Thevananther S, Kim C-S, et al. Redundant Pathways for Negative Feedback Regulation of Bile Acid Production. Developmental Cell. 2002;2:723–1. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 36.Huang J, Iqbal J, Saha PK, Liu J, Chan L, Hussain MM, et al. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46(1):147–57. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- 37.Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE, et al. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci U S A. 2006;103(22):8552–7. doi: 10.1073/pnas.0603115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Liu J, Saha P, Huang J, Chan L, Spiegelman B, et al. The orphan nuclear receptor SHP regulates PGC-1alpha expression and energy production in brown adipocytes. Cell Metab. 2005;2(4):227–38. doi: 10.1016/j.cmet.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 39.Park YJ, Kim SC, Kim J, Anakk S, Lee JM, Tseng HT, et al. Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J Lipid Res. 2011;52(12):2234–44. doi: 10.1194/jlr.M016048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suh YH, Kim SY, Lee HY, Jang BC, Bae JH, Sohn JN, et al. Overexpression of short heterodimer partner recovers impaired glucose-stimulated insulin secretion of pancreatic beta-cells overexpressing UCP2. J Endocrinol. 2004;183(1):133–44. doi: 10.1677/joe.1.05675. [DOI] [PubMed] [Google Scholar]
- 41.Edvardsson U, Bergstrom M, Alexandersson M, Bamberg K, Ljung B, Dahllof B. Rosiglitazone (BRL49653), a PPARgamma-selective agonist, causes peroxisome proliferator-like liver effects in obese mice. J Lipid Res. 1999;40(7):1177–84. [PubMed] [Google Scholar]
- 42.Kubota N, Terauchi Y, Kubota T, Kumagai H, Itoh S, Satoh H, et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281(13):8748–55. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- 43.Moore GB, Chapman H, Holder JC, Lister CA, Piercy V, Smith SA, et al. Differential regulation of adipocytokine mRNAs by rosiglitazone in db/db mice. Biochem Biophys Res Commun. 2001;286(4):735–41. doi: 10.1006/bbrc.2001.5460. [DOI] [PubMed] [Google Scholar]
- 44.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409(6818):307–12. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 45.Way JM, Gorgun CZ, Tong Q, Uysal KT, Brown KK, Harrington WW, et al. Adipose tissue resistin expression is severely suppressed in obesity and stimulated by peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2001;276(28):25651–3. doi: 10.1074/jbc.C100189200. [DOI] [PubMed] [Google Scholar]
- 46.Rajala MW, Qi Y, Patel HR, Takahashi N, Banerjee R, Pajvani UB, et al. Regulation of resistin expression and circulating levels in obesity, diabetes, and fasting. Diabetes. 2004;53(7):1671–9. doi: 10.2337/diabetes.53.7.1671. [DOI] [PubMed] [Google Scholar]
- 47.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278(36):34268–76. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, et al. Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J Biol Chem. 2006;281(49):37603–15. doi: 10.1074/jbc.M604709200. [DOI] [PubMed] [Google Scholar]
- 49.Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab. 2005;288(6):E1195–205. doi: 10.1152/ajpendo.00513.2004. [DOI] [PubMed] [Google Scholar]
- 50.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem. 2003;278(1):498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 51.Renga B, Mencarelli A, Migliorati M, Cipriani S, D'Amore C, Distrutti E, et al. SHP-dependent and -independent induction of peroxisome proliferator-activated receptor-gamma by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflamm Res. 2011;60(6):577–87. doi: 10.1007/s00011-010-0306-1. [DOI] [PubMed] [Google Scholar]
- 52.Kim SC, Kim CK, Axe D, Cook A, Lee M, Li T, et al. All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade. Hepatology. 2014;59(5):1750–60. doi: 10.1002/hep.26699. [DOI] [PMC free article] [PubMed] [Google Scholar]