

Abstract
The intestinal microbial ecosystem is complex, and few of the principles that contribute to homeostasis in health are well understood. Pham et al. (2014) show that a network including the epithelial interleukin-22 receptor protects against infection with the opportunistic pathogen Enterococcus faecalis through promotion of host-microbiota mutualism.
The human gastrointestinal (GI) tract is a rich and highly complex ecosystem, where a wide variety of bacterial, fungal, and viral inhabitants coexist with one other and in symbiosis with their host. This diverse microbial community aids digestion, synthesizes vitamins, and protects the host against infection. But the microbiota exists in a delicate balance that is periodically disrupted, resulting in a state of microbial dysbiosis. The factors that precipitate microbial dysbiosis, and how to prevent or reverse it, are not well understood.
Recent studies point to the complex interplay between diet, community complexity, and host immunity in maintaining GI tract microbial homeostasis (Turnbaugh et al., 2009; Jernberg et al., 2007; Deatherage Kaiser et al., 2013). Not surprisingly, diet is an important factor in determining GI community composition. When host diet shifts, the composition of the GI microbial ecosystem also shifts (Turnbaugh et al., 2009). Over the past 50 years, antibiotic treatment has emerged as an important cause of microbial dysbiosis; broad-spectrum antimicrobials fundamentally change the composition of GI tract flora, and the effects can persist for years (Jernberg et al., 2007). Infection with a virulent pathogen such as Salmonella can also alter community structure and result in dysbiosis (Deatherage Kaiser et al., 2013). Finally, the host immune system plays a key role in maintaining a microbial homeostasis compatible with health. The immune system is “trained” as healthy flora are established during development and becomes capable of distinguishing between beneficial and harmful GI tract microbes. When working properly, the immune system is able to promote the occurrence of the former and suppress the latter. But when this training goes awry, or when host immunity is disrupted, the GI tract ecosystem can tilt toward dysbiosis and disease (Figure 1).
Figure 1. Driving Forces in the Balance between Gastrointestinal Tract Homeostasis and Dysbiosis.
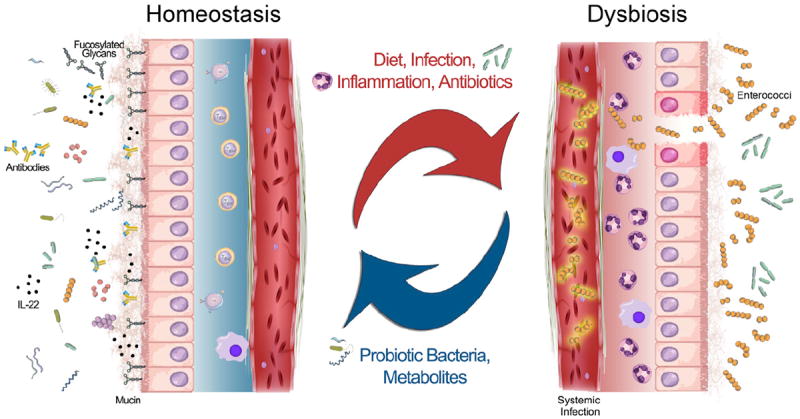
On the left, healthy GI tract homeostasis is characterized by a large diversity of microbes, an intact mucin layer, epithelial cells decorated with fucosylated glycans, and patrolling immune cells that produce antibodies, cytokines such as IL-22, and other antimicrobial peptides, all of which help regulate the microbial community. Disruptive forces such as changes in diet, infection with a virulent microbe, inflammation, and/or antibiotic use can shift the community to a state of dysbiosis, shown on the right. In this state, the microbial community is thrown off balance, and the host is prone to systemic infection with enterococci. Administering probiotic bacteria, or simply probiotic metabolites such as 2′-fucosyllactose, can help restore microbial homeostasis.
What are the key interactions between the host immune system and the GI tract microbial community that contribute to or prevent dysbiosis? In this issue, Pham and colleagues explore the role of the interleukin-22 receptor IL-22RA1 in maintaining microbial homeostasis in the mouse GI tract (Pham et al., 2014). Because intestinal inflammation can cause dysbiosis, the authors hypothesized that IL-22RA1 contributes to maintaining microbial homeostasis by restricting the overgrowth of opportunistic pathogens. In a series of experiments in mice lacking IL-22RA1, the authors demonstrate that during microbial dysbiosis, the opportunistic pathogen Enterococcus faecalis is able to expand in relative abundance in the intestinal tract and then translocate to the bloodstream and other organs. The authors show that signaling through IL-22RA1 results in induction of the fucosyltransferase Fut2, which helps restore community diversity, possibly through increased availability of fucosylated oligosaccharides such as 20-fucosyllactose. Another recent study found that fucosylation of intestinal epithelial cells is regulated by Fut2 and depends upon the interleukin-22 (IL-22) cytokine (Goto et al., 2014). Collectively, these findings lead to a model whereby IL-22, IL-22RA1, and Fut2 help maintain host-microbiota mutualism by promoting the growth of probiotic anaerobic bacteria belonging to the Ruminococcus and Bacteroides genera, which thrive on fucosylated glycans (Figure 1). Promoting the growth of these commensal anaerobes would limit the overgrowth of E. faecalis, thus maintaining microbial homeostasis.
The selective outgrowth of enterococci following intestinal dysbiosis is emerging as a common theme, regardless of whether dysbiosis results from antibiotic treatment, intestinal inflammation, or infection. Administration of broad-spectrum antimicrobials decreases community diversity and is associated with intestinal dominance and systemic infection with enterococci (Ubeda et al., 2010). Antibiotic treatment also eliminates microbe-derived products, such as the flagella of Gram-negative organisms, from the intestinal niche. Normally, these microbe-derived products stimulate secretion of host defense molecules, such as RegIIIγ, into the mucosa. With reduced antimicrobial RegIIIγ expression, enterococci are able to overgrow in the intestine and ultimately invade the bloodstream (Kinnebrew et al., 2010). Furthermore, a recent study of microbiota changes following intestinal surgery found that anastomotic injury, and presumably the resulting inflammation, caused a 500-fold increase in the relative abundance of enterococci in the intestinal tissue-associated microbiota (Shogan et al., 2014). Finally, infection of mice with Salmonella enterica serovar Typhimurium has been found to induce GI tract inflammation and microbial dysbiosis and again lead to increased abundance of enterococci (Deatherage Kaiser et al., 2013). The commonality between these studies is the elimination of probiotic commensal microbes and the selective expansion of enterococci to high density, presumably because of the lack of competition for nutrients and space.
The enterococci belong to an ancient genus that we believe arose with the development of the microbiota in complex life forms and has evolved along with the tree of life (Van Tyne and Gilmore, 2014). Today, enterococci are found in a wide range of hosts that spans the animal kingdom, from insects to man. Two Enterococcus species, E. faecalis and E. faecium, have become well-adapted hospital pathogens and are common causes of antibiotic-resistant infection. E. faecalis and to a lesser extent E. faecium are also core members of the healthy human GI tract microbiota, and for this reason, when they overgrow and infect ordinarily sterile sites, they are referred to as opportunistic pathogens. Enterococci can cause urinary tract infections, endocarditis, sepsis, and wound infections; these infections are particularly difficult to treat because of intrinsic antibiotic resistance, stemming from the inherent ruggedness of the bacteria and additional resistances that have been acquired on mobile genetic elements.
What unique features allow enterococci to proliferate when microbial homeostasis is disrupted? In addition to intrinsic and acquired antibiotic resistances, E. faecalis and E. faecium are able to resist other antimicrobial factors, such as bile, and are able to tolerate a wide variety of stressors that would eliminate their competitors. Furthermore, enterococci are able to persist in the environment longer than other bacteria; if they were similarly able to persist longer in the perturbed GI tract, they would gain access to a competitor-free niche, where they could resume growth when the opportunity arises. While these and other factors likely contribute to the proliferation of enterococci during GI tract dysbiosis, many of the mechanistic details underlying this phenomenon remain to be discovered.
What can be done to reverse or prevent microbial dysbiosis and thus prevent subsequent enterococcal infections? Maintaining a diverse GI tract microbial community appears to be crucial, but how can diversity be restored therapeutically? On an ecosystem level, transplantation of normal microbial flora from a healthy donor has been used successfully to manage GI tract ecology and restore population diversity in patients suffering from chronic infection with Clostridium difficile (Lo Vecchio and Cohen, 2014). With specific reference to limiting enterococcal overgrowth, activation of the IL-22 receptor is a theoretical possibility but is complicated by the fact that the receptor occurs on a variety of cell types and is connected to multiple signaling networks. Restoring community diversity with obligate anaerobes, such as Barnsiella, Bacteroides, or Ruminococcus, could also be worthwhile. The findings of Pham and colleagues, however, suggest a direct and simpler approach: administration of fucosylated oligosaccharides, such as 2′-fucosyllactose, which may do much to restore community complexity and promote homeostasis.
Acknowledgments
The authors wish to thank José T. Saavedra for assistance in developing the figure that accompanies this article. For work on enterococcal colonization, virulence, and antibiotic resistance, the authors gratefully acknowledge the support of NIH grants AI072360 and AI108710, the Harvardwide Program on Antibiotic Resistance AI083214, and fellowship support from Grant AI109855 (to D.V.T.).
References
- Deatherage Kaiser BL, Li J, Sanford JA, Kim YM, Kronewitter SR, Jones MB, Peterson CT, Peterson SN, Frank BC, Purvine SO, et al. PLoS ONE. 2013;8:e67155. doi: 10.1371/journal.pone.0067155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, Takeyama N, Kamioka M, Sakamoto M, Matsuki T, et al. Science. 2014;345:1254009. doi: 10.1126/science.1254009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernberg C, Löfmark S, Edlund C, Jansson JK. ISME J. 2007;1:56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- Kinnebrew MA, Ubeda C, Zenewicz LA, Smith N, Flavell RA, Pamer EG. J Infect Dis. 2010;201:534–543. doi: 10.1086/650203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Vecchio A, Cohen MB. Curr Opin Gastroenterol. 2014;30:47–53. doi: 10.1097/MOG.0000000000000023. [DOI] [PubMed] [Google Scholar]
- Pham TAN, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, Keane JA, Page AJ, Kumasaka N, Kane L, et al. Cell Host Microbe. 2014;16:504–516. doi: 10.1016/j.chom.2014.08.017. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogan BD, Smith DP, Christley S, Gilbert JA, Zaborina O, Alverdy JC. Microbiome. 2014;2:35. doi: 10.1186/2049-2618-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, Pamer EG. J Clin Invest. 2010;120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tyne D, Gilmore MS. Annu Rev Microbiol. 2014;68:337–356. doi: 10.1146/annurev-micro-091213-113003. [DOI] [PMC free article] [PubMed] [Google Scholar]