

Abstract
The purpose of this study was to evaluate the integrity of the muscle membrane and its associated cytoskeleton after a contraction-induced injury. A single eccentric contraction was performed in vivo on the tibialis anterior (TA) of male Sprague-Dawley rats at 900°/s throughout a 90°-arc of motion. Maximal tetanic tension (Po) of the TAs was assessed immediately and at 3, 7, and 21 days after the injury. To evaluate sarcolemmal integrity, we used an Evans blue dye (EBD) assay, and to assess structural changes, we used immunofluorescent labeling with antibodies against contractile (myosin, actin), cytoskeletal (α-actinin, desmin, dystrophin, β-spectrin), integral membrane (α- and β-dystroglycan, sarcoglycan), and extracellular (laminin, fibronectin) proteins. Immediately after injury, P0 was significantly reduced to 4.23 ± 0.22 N, compared with 8.24 ± 1.34 N in noninjured controls, and EBD was detected intra-cellularly in 54 ± 22% of fibers from the injured TA, compared with 0% in noninjured controls. We found a significant association between EBD-positive fibers and the loss of complete dystrophin labeling. The loss of dystrophin was notable because organization of other components of the subsarcolemmal cytoskeleton was affected minimally (β-spectrin) or not at all (α- and β-dystroglycan). Labeling with specific antibodies indicated that dystrophin’s COOH terminus was selectively more affected than its rod domain. Twenty-one days after injury, contractile properties were normal, fibers did not contain EBD, and dystrophin organization and protein level returned to normal. These data indicate the selective vulnerability of dystrophin after a single eccentric contraction-induced injury and suggest a critical role of dystrophin in force transduction.
Keywords: muscle injury, dystrophin, cytoskeleton, sarcolemma
Skeletal muscle injury is characterized by an immediate loss of the ability to produce force. The cause of this force loss has been attributed to such factors as a defect in excitation-contraction (EC) coupling (55), disruption or loss of force-generating structures such as actin and myosin (49), and disruption or loss of force-transmitting structures, such as desmin (6, 31). Perhaps the best evidence that disruption of force-bearing structures contributes to strength loss after injury comes from single-fiber studies, where reduction in single-fiber maximal force is observed immediately after eccentric injury (32, 33). Although the totality of injury is likely the result of many factors, the purpose of this study was to assess structural defects of the sarcolemma, dystrophin, and dystrophin-associated proteins after a single traumatic muscle injury and to relate these observations to contractile properties.
The sarcolemma transmits force and is subjected to substantial stress during contractions (48). The sarcolemma is linked to the contractile apparatus by a membrane-associated cytoskeleton, which contains proteins that have also been implicated in force transmission and have been described in several reviews (41, 50). Damage to the sarcolemma and its associated cytoskeleton has been shown to occur after suspension reloading (52) and repeated eccentric contractions (20, 37). The significance of cytoskeletal proteins is underscored by genetic defects in one or more of these proteins, as seen clinically in muscular dystrophies in humans, such as Duchenne’s or Becker’s (deleted or truncated dystrophin, respectively) or Limb Girdle (mutations in α- and γ-sarcoglycan) dystrophies, and in mice lacking dystrophin (mdx) (15, 19, 43). In all these conditions, a defect in the membrane cytoskeleton gives rise to histopathological changes, which are associated decreased muscle performance.
Several outstanding studies in recent years have focused on the contribution of mechanical factors in eccentric contraction-induced muscle injury (9, 17, 24, 30, 38, 54); however, to date there is limited information on the cellular consequences of a single eccentric contraction-induced injury. The importance of the sarcolemma-associated cytoskeleton is highlighted by the presence of pathological findings seen with dystrophies, but few studies are available that directly associate contractile function with structural changes at the membrane and its associated cytoskeleton. In this study, we performed a controlled single eccentric contraction of rat tibialis anterior muscles (TAs) and assessed the consequences at the level of the membrane and its associated cytoskeleton. The goal was to induce a mechanical perturbation that would have a selective loss of certain proteins. We hypothesized that dystrophin, a putative link in lateral transduction of mechanical force, would be selectively affected by the eccentric contraction-induced injury. We also tested whether the contractile function and membrane integrity would recover after the eccentric contraction-induced injury.
METHODS
Muscle injury
Animal use was approved by the University of Maryland Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (n = 30; Charles River Laboratories, Wilmington, MA), weighing 386 ± 42 g (age 3–4 mo), were randomly distributed into one of five groups: noninjury controls (NI, n = 6), injury day 0 (D0, n = 6), injury day 3 (D3, n = 6), injury day 7 (D7, n = 6), and injury day 21 (D21, n = 6). The rats were anesthetized with intraperitoneal ketamine/xylazine (40:10 mg/kg body mass, respectively). With the animal supine, the hindlimb was stabilized using a transosseous 16-gauge needle through the tibia. The foot was secured to a plate attached to a stepper motor (model T8904; NMB Technologies, Chatsworth, CA). The peroneal nerve was dissected free through a small incision and clamped with a subminiature electrode (Harvard Apparatus, Holliston, MA) that was used to stimulate the TA with a supramaximal tetanic current (75-Hz, 300-ms train duration with 1-ms pulses at a constant current of 5 mA). We used 150% of the maximum stimulation intensity to activate the TA in our experiments to induce maximal contractile activation. An isolation unit (model PSIU6; Grass Instruments, Warwick, RI) was used between the stimulator and electrode to minimize artifact. Our protocol used commercial software (Labview version 4.1; National Instruments, Austin, TX) to independently control the onset of contractile activation, angular velocity, and arc of motion during plantar flexion.
The eccentric contraction was performed through a 90° arc of motion at an angular velocity of 900°/s, starting with the foot orthogonal to the tibia. The TA was stimulated 200 ms isometrically prior to and throughout the arc of motion. NI were TAs that underwent the same procedure and were passively lengthened throughout the range without the eccentric contraction.
Contractile function
Contractile properties of all TAs were obtained within minutes (NI and D0) or at specific time points (D3, D7, D21) after the perturbation. The distal tendon of the TA was released, and its proximal portion was secured in a custom-made metal clamp and attached to a load cell (FT03; Grass instruments) using a suture tie (4.0 coated Vicryl). The load cell was mounted to a micromanipulator (Kite Manipulator; World Precision Instruments, Sarasota, FL) so that the TA could be adjusted to resting length. The tibia was stabilized with a 16-gauge needle, and the peroneal nerve was used to stimulate the TA. The TA was protected from cooling by a heat lamp and from dehydration by mineral oil. Tetanic force (achieved by a train duration of 300 ms with 1-ms square pulses at 75 Hz) was recorded, and the signal from the load cell was fed via a DC amplifier (model P122; Grass Instruments) to an analog-to-digital board using acquisition software (PolyVIEW version 2.1; Grass Instruments).
Injection of Evans blue dye
To study the integrity of the muscle fiber membrane, animals (3 per experimental group) received an intraperitoneal injection of 1% Evans blue dye (EBD; Sigma, St. Louis, MO) (wt/vol) in phosphate-buffered saline (PBS, pH 7.4) at a volume of 1% body mass (BM) (1 mg EBD/0.1 ml PBS/10 g BM). This solution was sterilized by passage through a Millex-GP 0.22 μm filter (Millipore, Bedford, MA) and administered 24 h before death of the animal to assure a good signal. EBD binds to albumin and is detected by fluorescence microscopy (at 568 nm) in the extracellular space. Presence of the protein-bound dye inside the muscle fiber (Fig. 4A) indicates damage to the sarcolemma (20, 36, 47).
Fig. 4.

Double labeling of dystrophin and desmin. Fluorescent confocal images (×25) from double labeling of dystrophin and desmin of uninjured (A–C) and injured (D–F) tissue sections. Sections were labeled with monoclonal antibodies against dystrophin (B and E and light blue in overlay C or yellow/green in overlay F) and polyclonal antibodies against desmin (A and D and blue in C and F). In noninjured muscle, desmin labeling is throughout the fibers and dystrophin labeling is circumferential as expected. In injured fibers, loss of desmin labeling is more diffuse (asterisk in D), and there is incomplete labeling of dystrophin (arrow in E). Scale bar = 25 μm.
Cryosectioning
Animals were anesthetized, and the TAs were harvested either after perfusion fixation with 4% paraformaldehyde (EBD injected, n = 3 per experimental group) or before death without fixation (for Western blots, n = 3 per experimental group). To study morphology, the TA was cut into a proximal, middle, or distal third, and frozen tissue was cryosectioned (10 μm thickness). The sections were collected onto slides coated in a 2% solution of organosilane (3-aminopropyltriethoxy; Sigma).
Antibodies
Monoclonal antibodies to dystrophin (NCL-dys-1 and dys-2), myosin (WB-MHCf), and α- and β-dystroglycan were purchased from Novocastra (Newcastle upon Tyne, UK). Monoclonal antibodies to actin, α-actinin, desmin, and laminin were purchased from Sigma. Polyclonal antibodies to β-spectrin were generously provided by Dr. Robert Bloch (University of Maryland, Baltimore, MD). Secondary antibodies included donkey anti-mouse, goat anti-mouse, and goat anti-chicken, conjugated to fluorescein isothiocyanate (FITC) for use in immunofluorescence experiments, or goat anti-mouse conjugated to alkaline phosphatase for use in immunoblotting. The secondary antibodies were from Jackson Immunoresearch Laboratories (West Grove, PA) and were species-specific with minimal cross reactivity.
Fluorescence imaging and immunolabeling
To quantify the percentage of EBD-positive fibers, EBD was visualized by fluorescence microscopy of the cryosections. A muscle fiber that contained intra-cellular EBD was considered positive independent of the amount of dye that was present. During a pilot study, we noted that the EBD signal was present throughout the muscle, including the myotendinous junction; however, the highest number of positive fibers was found in the middle third. Sections were randomized and viewed at ×25. Each optical field contained an average of 42 ± 4.6 fibers, and >20 fields were counted per muscle third. The EBD data are presented as the total number of EBD-positive fibers per TA at each time point. For the immunohistochemistry, only the middle third of each TA was used from NI, D0, and D21. We examined sections for the presence of EBD at various time points after the eccentric contraction (D0, D3, D7, D21) and from NI.
Sections with EBD-positive fibers were also labeled with antibodies against laminin (1:500), α-dystroglycan (1:200), and β-dystroglycan (1:200), as well as dystrophin (dys2, 1:10; dys1, 1:10), β-spectrin (3 ug/ml), and desmin (1: 50). Sections were washed in 100 mM glycine/PBS for 10 min, permeabilized in 0.5% Triton/PBS for 10 min, and blocked in BSA/PBS (1 mg/ml) for 1 h. They were then incubated in primary antibody for 1 h at room temperature. After being washed with BSA/PBS, the samples were incubated with the appropriate FITC- or Cy5-conjugated secondary antibody for 1 h. The excess secondary was washed off with PBS, and the sections were mounted in 90% glycerol, 1 M Tris·HCl, pH 8.0, and supplemented with 1 mg/ml p-phenylenediamine to reduce fading. A Zeiss 410 confocal laser-scanning microscope was used to obtain images of sections. All labeling was shown to be specific by use of the appropriate nonimmune sera. The digital images were not processed by imaging software.
SDS-PAGE and immunoblotting
Western blot analysis was used to assess semiquantitative changes in the levels of representative muscle proteins. The middle third of unfixed tissue was cooled in liquid nitrogen, pulverized, and then homogenized in Laemmli buffer. Samples were boiled and centrifuged, and the protein concentration of the supernatant was determined using an amido black assay (21). Samples were then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose electrophoretically. The nitrocellulose was blocked overnight in 3% milk-PTA (0.1% Tween/PBS, 10 mM sodium azide), washed, and then incubated with the monoclonal antibody for 2 h. We used antibodies against dystrophin (dys2), desmin, and α-actinin. Excess antibodies were washed off, and the nitrocellulose incubated with donkey anti-mouse secondary antibodies was conjugated to alkaline phosphatase (Jackson Laboratories). The excess secondary antibodies were then washed off with 0.1% polyoxethylene sorbitan/PBS, and the bands were visualized by a chemiluminescent assay method (Tropix, Bedford, MA).
Statistics
Contractile data from each experiment were analyzed using a single factor analysis of variance (ANOVA, SigmaStat, San Rafael, CA), with Po as the dependent variable. When a significant F-ratio was found, a Tukey post hoc analysis was performed to determine where significant differences had occurred. Power was >0.8. To analyze the relationship between EBD-positive fibers and dystrophin-disrupted fibers, a χ2 test was used with on a 2 × 2 contingency table. Significance was set at P < 0.05, and all results are reported as means ± SD.
RESULTS
Muscle contractile function and membrane damage
To evaluate the loss of contractile function after a single eccentric contraction-induced injury, we measured maximal tetanic tension (Po) within 15 min after the injury (day 0) and at days 3, 7, and 21 (Fig. 1B). In noninjured controls, Po was 8.24 ± 1.34 N, and within 15 min after the eccentric contraction, maximal tetanic tension decreased to 4.23 ± 0.22 N. Tetanic tension continued to decrease (2.83 ± 1.03 N at day 3). This further decline in muscle force has been noted in other studies and has been termed a “secondary injury” (21), likely due to inflammation (34, 35, 46, 51, 57). Standard histological staining (hematoxylin and eosin) of sections 1 day after the eccentric contraction showed signs of a robust response, with numerous cells migrating from the vasculature into the extracellular space (data not shown), indicative of an inflammatory response. One week after the injury tetanic tension increased to 6.07 ± 1.23 N and by day 21, tetanic tension was not different from noninjured controls (9.31 ± 1.18 N). To assess the degree of membrane damage, we counted the muscle fibers that contained intracellular EBD in each muscle. Within 15 min of the eccentric contraction, 54 ± 9% of the fibers in the TA contained the dye (see Fig. 1, A and B). The percentage of EBD-positive muscle fibers on days 3, 7, and 21 was 28 ± 6%, 8 ± 6%, and 2 ± 2%, respectively. In noninjured TAs, EBD was excluded form the muscle fibers. The data demonstrate the complementary changes between maximal tetanic tension and EBD-positive muscle fibers. The experiments furthermore indicate that the eccentric contraction-induced injury led to a recovery of contractile function, which was associated with the integrity of the membrane.
Fig. 1.

Membrane damage with loss of function. A: integrity of the sarcolemma was assessed by permeation of Evans blue dye (EBD). Noninjured TAs did not show dye uptake into the fibers (left), whereas injured TAs did (right), indicating sarcolemma damage. B: the percentage of EBD-positive fibers (dashed line) is plotted against contractile function (solid line), as measured by maximal isometric tetanic tension (Po). EBD was found in a high percentage of fibers immediately after the injury, when Po dropped significantly from noninjured values. Minimal EBD was seen 21 days postinjury, when contractile function was fully restored.
Immunofluorescent labeling
To further characterize the nature of the membrane damage by the eccentric contraction, we used immunofluorescent labeling of extracellular, integral membrane, membrane-associated cytoskeletal, and contractile proteins from samples obtained within minutes after the eccentric contraction. We used antibodies against laminin, α-dystroglycan, and β-dystroglycan, as well as dystrophin, β-spectrin, and desmin. Double immunofluorescent labeling allowed us to visualize EBD-positive fibers with laminin (Fig. 2, A–C), α-dystroglycan (Fig. 2, D–F), or β-dystroglycan (Fig. 2, G–I). We found that fibers positive for EBD had intact organization of laminin, α-dystroglycan, and β-dystroglycan in all the sections that we evaluated. In a second series of experiments, we used the same technique and saw that dystrophin organization was lost at the membrane of EBD positive muscle fibers (Fig. 3, A–C). In fibers of injured muscle that did not contain EBD, the dystrophin labeling was consistently intact. Another component of the membrane-associated cytoskeleton (β-spectrin, Fig. 3, D–F) was affected, but not as pronounced, and fluorescent labeling of desmin (Fig. 3, G–I) showed a diffuse loss within the muscle fiber (Fig. 3, G–I, arrows). We found a significant association between EBD-containing muscle fibers and the loss of dystrophin (P < 0.001). In experiments in which we used antibodies against contractile molecules (sarcomeric actin and myosin heavy chain) on tissue cross sections, we did not observe any changes suggesting a loss of these molecules in fibers that were positive for EBD (data not shown). The data suggest that in this model of injury, dystrophin is selectively more affected by the single eccentric contraction.
Fig. 2.

Labeling of membrane-associated cytoskeleton after injury. Tissue sections from injured tibialis anterior muscles (TAs) were labeled for laminin (A–C), α-dystroglycan (D–F), and β-dystroglycan (G–I). The left column shows images of the respective molecules of interest. EBD-positive fibers from the same section are shown in the middle column, and the right column shows the overlay of the EDB (red) with fluorescent labeling (green). EBD was administered by intraperitoneal injection 24 h before injury. The data indicate that the organization of membrane-associated molecules is not affected. Scale bar = 50 μm.
Fig. 3.

Labeling of the subsarcolemmal cytoskeleton after injury. Tissue sections from injured TAs were labeled for dystrophin (AC), β-spectrin (D–F), and desmin (G–I). The left column shows images of the respective molecules of interest. EBD-positive fibers from the same section are shown in the middle column, and the right column shows the overlay of the EDB (red) with fluorescent labeling (green). EBD was administered by intraperitoneal injection 24 h before injury. Fibers that were permeable to EBD also lacked dystrophin (arrows, A–C). β-Spectrin and desmin were much less affected in injured fibers (arrows, D–F and G–I, respectively). Scale bar = 50 μm.
To discern in more detail the changes in the membrane-associated cytoskeleton, we performed double immunofluorescent labeling with antibodies against desmin and the COOH terminus of dystrophin (dys2 MAb). The normal relationship is shown in a representative image from a noninjured control (Fig. 4, A–C). The eccentric contraction-induced injury led to a pronounced focal loss of dystrophin (arrows in Fig. 4, E and F) compared with desmin, which showed a loss that was more diffuse and intracellular (asterisks in Fig. 4, D and F). We also labeled sections from the EBD-injected animals with monoclonal antibodies that bind only to the rod domain (dys1 MAb) of the dystrophin molecule to detect any selective loss of labeling (Fig. 5). The epitopes recognized by these different monoclonal antibodies have been located with high resolution by biochemical and recombinant DNA mapping techniques (14, 22). Using serial sections, our data suggested that whereas the rod domain of dystrophin was typically present at the membrane (Fig. 5A), the COOH-terminal end of the molecule was not visible (Fig. 5B). Together, the experiments indicate that within 15 min after the eccentric contraction, the intracellular organization of desmin may be affected and the organization of dystrophin is preferentially lost, especially at the COOH-terminal end of the molecule.
Fig. 5.
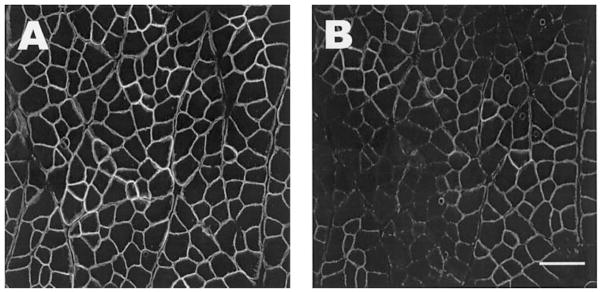
Loss of dystrophin labeling is confined to the COOH terminus. Serial sections from an injured TA are labeled with monoclonal antibodies to either the rod domain (dys1, A) or the COOH terminus (dys2, B), showing a selective loss of labeling to the COOH terminus. Scale bar = 50 μm.
Recovery and Western blot analysis
To obtain a semiquantitative analysis of our findings, we performed Western blot analysis with antibodies against dystrophin (dys2), desmin, and α-actinin (a representative experiment is shown in Fig. 6). The relative amounts of protein were determined by the densitometric signal of the bands on days 0, 3, 7, and 21 and converted to the percentage of the protein levels of noninjured TAs, which were taken as 100%. A Coomassie stain of representative samples indicated that no major protein degradation was present and that all lanes contained equal amounts of protein (not shown). As expected, we saw that dystrophin was lost compared with desmin and α-actinin. Even though there was modulation in the amount of desmin and α-actinin over the different time points, it was not significant. In an additional experiment, we evaluated the dystrophin cytoskeleton 21 days after the injury and saw that 99 ± 3% of the muscle fibers had a dystrophin organization that was similar to noninjured muscles. The Western blots of dystrophin showed that after an initial loss of 68 ± 22% compared with noninjured controls, the levels of dystrophin returned to normal levels by day 21 and, thus, paralleled the recovery of contractile function and membrane integrity.
Fig. 6.

Western blot analysis of dystrophin and desmin from injured TA muscles. Western blot analysis was performed on injured TA at various time points after injury. NI are noninjured TAs, and D0 represents TAs harvested within 15 min postinjury. Data are also included for 3, 7, and 21 days after injury (D3, D7, D21, respectively). Each lane contains 20 μg of protein. A significant drop in the chemiluminescent signal is seen for dystrophin after injury (A). Although modulated, desmin does not show the same loss (B). The nonmembrane protein α-actinin was used as a control (C). Each bar represents data from 3 independent experiments, and the histograms represent means ± SD.
DISCUSSION
The present study examined the extent of membrane and cytoskeletal damage in skeletal muscle fibers after a single eccentric contraction. The findings indicate that within minutes there is a loss of contractile function, membrane integrity, and dystrophin, especially its COOH-terminal end. The organization of dystrophin was selectively affected, whereas that of its associated molecules, α- and β-dystroglycan, was not. The organization of β-spectrin and desmin was affected to a much lesser extent compared with dystrophin. The dystrophin protein levels, as well as its submembrane organization and contractile function, returned to preinjury status 21 days after the eccentric contraction-induced injury. Our experiments underline the importance of sarcolemmal integrity and contractile function and point to a selective vulnerability of dystrophin in eccentric contraction-induced injury.
A loss of cytoskeletal protein labeling was localized primarily to dystrophin, a large subsarcolemmal protein concentrated at the Z and M lines and thought to be important for force transmission (41, 42, 50). The reduction or loss of dystrophin was a prominent finding in fibers that sustained sarcolemmal damage, as indicated by the significant association (χ2) between muscle fibers absent for dystrophin and muscle fibers positive for intracellular EBD (47). We did not see a uniform loss of desmin, which may be the result of a smaller extent of mechanical perturbation using a single eccentric contraction compared with repetitive eccentric contractions, which result in a uniform loss of desmin (26) or from inflammatory processes (46). However, the loss of a molecule thought to be involved in force transduction may paradoxically have a protective effect (20, 37, 44, 52). Serum enzymes, such as creatine kinase (CK-MM), have frequently been used as a marker of sarcolemma damage (10, 27). However, enzyme efflux does not correlate well with observed pathological changes or loss of contractile function (18, 27, 39).
Disruption of both the sarcolemma and internal cytoskeleton has previously been implicated in loss of force production after muscle injury (20, 37, 44, 51). Force is generated within sarcomeres, and it is generally accepted that this force is transmitted longitudinally along the myofibril (41). However, there is evidence that force is also transmitted radially through intermediate filaments (e.g., desmin) of the internal cytoskeleton and outward toward the sarcolemma (41, 48). By imposing an eccentric contraction, the high forces generated may strain components of the putative force-transduction pathway. Our results suggest that dystrophin plays an important role in that pathway. Integral membrane proteins of the dystrophin complex (α- and β-dystroglycan) were not affected by the eccentric contraction, although β-spectrin and desmin showed a smaller degree of disorganization, suggesting that dystrophin is likely to be the most vulnerable link in the transduction of lateral force (Fig. 7).
Fig. 7.
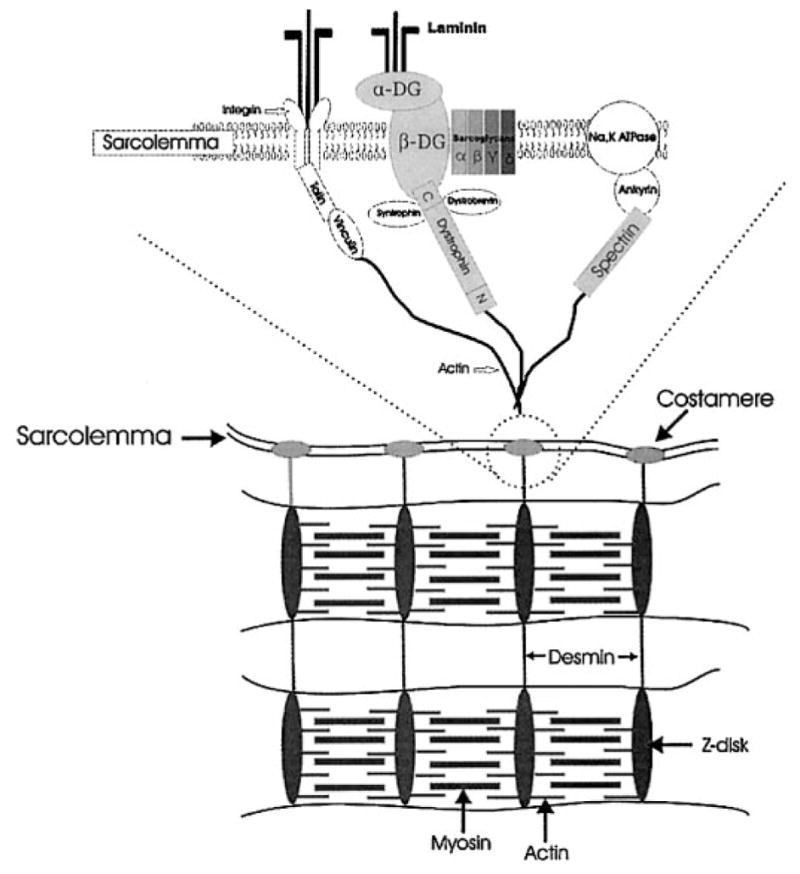
Schematic of selected membrane-associated molecules.
On the other hand, the loss of dystrophin organization may not be solely due to a direct mechanical disruption of elements supposedly responsible for force transduction. There have been reports that dystrophin is vulnerable to calcium-activated pro-teases (1, 13, 45, 53). Because we show in our model of muscle injury that the integrity of the membrane is seriously compromised, it is reasonable to assume that extracellular calcium enters the muscle fiber. In the present study, we found that the loss of dystrophin labeling immediately after injury was confined to the COOH terminus of this large molecule. This finding may be the result of unique proteinase-sensitive cleavage sites along the molecule. In fact, certain viruses produce proteases that target dystrophin (4, 5). Proteinase-sensitive regions have been identified in dystrophin isolated from several species, including humans (25, 56), and studies of dystrophin in situ indicate that there are proteinase-resistant regions in the rod domain (3). The NH2-terminal half of dystrophin is much more protected from digestion (23), possibly because it binds to actin, which might provide steric protection from proteases. In a cell-free system, the COOH terminus of dystrophin is rapidly and preferentially degraded by calpains (13). The COOH terminus is likely to be important because its deletion causes severe muscular dystrophy (7, 43), and this is the region typically affected in Duchenne’s muscular dystrophy (12). The current study found that the COOH-terminal region of dystrophin was preferentially affected soon after injury. However, at this point, we cannot rule out that some of the degradation may be a result of the proteolytic activity of the inflammatory cells associated with the injury. Furthermore, despite the return of dystrophin protein levels, its organization, and the return of contractile function 21 days after the injury, we cannot directly correlate the contractile function with the return of dystrophin organization because we did not quantify the subsarcolemmal structures at several time points. Finally, we used an anesthetic that may slightly suppress the development of tetanic force (28) and may limit the interpretation of our results.
The rapid loss of dystrophin seen in the present study is consistent with models of force transduction within fibers (16, 40, 42), as well as findings from other studies that examine cytoskeletal changes (2, 8, 11, 26). A rapid and specific loss of dystrophin labeling has also been reported in ischaemically injured cardiomyocytes without affecting dystrophin-associated proteins, such as α-sarcoglycan, β-dystroglycan, and the Na+/Ca2+ exchanger membrane-protein (2, 7). A selective loss of dystrophin (30%) without concomitant loss of other cytoskeletal proteins such as desmin or α-actinin has also been reported after using a hindlimb suspension-reloading model in rats (11). More severe disruptions of dystrophin, including α-sarcoglycan, β-dystroglycan, and γ-sarcoglycan, have been shown in severe and vigorous exercise regimens. It is important to note that contractile function was not assessed in any of the above studies, and it is unclear whether the altered morphology was associated with decreased contractile function.
In the present study, we infer that the fibers with membrane damage and cytoskeletal disruption recovered from the injury because 21 days after the injury we observed that the organization of dystrophin was normal, as was contractile function. At this point, we have no direct evidence that the gradual reconstitution of the dystrophin complex is directly related to the return of contractile function, but the data show that the return of full contractile function corresponded to decreased membrane permeability. Although dystrophin loss was a paramount finding, our data show that there was not a concomitant loss of other dystrophin-associated proteins or internal molecules, suggesting that dystrophin may be a critical link in mechanical force transduction. Dystrophin’s binding partners, such as β-dystroglycan or β-spectrin, may form the next link in the putative pathway of radial force transmission, but our data suggest that they are not as vulnerable as dystrophin to either mechanical disruption or protein degradation. Desmin is an intracellular protein thought to link individual myofibrils laterally to each other and is lost after repeated eccentric contractions (31). In that study, the extensor digitorum longus (EDL) had an immediate loss of desmin, and it was only after 15 min of repeated contractions that the TA showed a significant loss in desmin, indicating that anatomical and biomechanical variables are likely important. Like muscles in dystrophin-knockout mice, muscles in desmin-knockout mice are reported to be more vulnerable to eccentric injury (29), although a different study indicates that muscles in desmin-knockout mice are much less vulnerable to injury than controls (44). Such disparate findings illustrate the present lack of full understanding we have for these various cytoskeletal proteins. The organization of desmin was affected by the traumatic injury we imposed, but our findings suggest it was limited to a small number of fibers.
In this study, we performed a single maximal eccentric contraction that resulted in injury and assessed the consequences at the level of the membrane and its associated cytoskeleton. The findings illustrate a complementary role between contractile function and membrane integrity. Moreover, our experiments suggest that the COOH terminus of dystrophin is preferentially lost quite soon after a single eccentric contraction-induced injury and that a 21-day period of postinjury recovery allows the dystrophin protein level and its submembrane organization to return to the preinjury status. Our experiments underline the importance of dystrophin in lateral force transduction and contractile function, but future experiments that focus on the reconstitution of the dystrophin complex will help to further elucidate mechanism of force transduction.
Acknowledgments
We thank Dr. Robert Bloch and Dr. David Pumplin for insight and contributions.
GRANTS
This work was funded in part by National Institutes of Health Grants T-32-AR-07592-06 (to R. M. Lovering) and K-01-HD-01165 (to P. G. De Deyne) and the National Football League Charities (to P. G. De Deyne).
References
- 1.Alderton JM, Steinhardt RA. How calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. Trends Cardiovasc Med. 2000;10:268–272. doi: 10.1016/s1050-1738(00)00075-x. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong SC, Latham CA, Shivell CL, Ganote CE. Ischemic loss of sarcolemmal dystrophin and spectrin: correlation with myocardial injury. J Mol Cell Cardiol. 2001;33:1165–1179. doi: 10.1006/jmcc.2001.1380. [DOI] [PubMed] [Google Scholar]
- 3.Augier N, Leger J, Robert A, Pons F, Leger JJ, Mornet D. Proteolytic susceptibility of the central domain in chicken gizzard and skeletal muscle dystrophins. Biochim Biophys Acta. 1992;1138:297–304. doi: 10.1016/0925-4439(92)90007-a. [DOI] [PubMed] [Google Scholar]
- 4.Badorff C, Berkely N, Mehrotra S, Talhouk JW, Rhoads RE, Knowlton KU. Enteroviral protease 2A directly cleaves dystrophin and is inhibited by a dystrophin-based substrate analogue. J Biol Chem. 2000;275:11191–11197. doi: 10.1074/jbc.275.15.11191. [DOI] [PubMed] [Google Scholar]
- 5.Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5:320–326. doi: 10.1038/6543. [DOI] [PubMed] [Google Scholar]
- 6.Barash IA, Peters D, Friden J, Lutz GJ, Lieber RL. Desmin cytoskeletal modifications after a bout of eccentric exercise in the rat. Am J Physiol Regul Integr Comp Physiol. 2002;283:R958–R963. doi: 10.1152/ajpregu.00185.2002. [DOI] [PubMed] [Google Scholar]
- 7.Bies RD, Caskey CT, Fenwick R. An intact cysteine-rich domain is required for dystrophin function. J Clin Invest. 1992;90:666–672. doi: 10.1172/JCI115909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biral D, Jakubiec-Puka A, Ciechomska I, Sandri M, Rossini K, Carraro U, Betto R. Loss of dystrophin and some dystrophin-associated proteins with concomitant signs of apoptosis in rat leg muscle overworked in extension. Acta Neuropathol (Berl) 2000;100:618–626. doi: 10.1007/s004010000231. [DOI] [PubMed] [Google Scholar]
- 9.Brooks SV, Zerba E, Faulkner JA. Injury to muscle fibres after single stretches of passive and maximally stimulated muscles in mice. J Physiol. 1995;488:459–469. doi: 10.1113/jphysiol.1995.sp020980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon JG, Orencole SF, Fielding RA, Meydani M, Meydani SN, Fiatarone MA, Blumberg JB, Evans WJ. Acute phase response in exercise: interaction of age and vitamin E on neutrophils and muscle enzyme release. Am J Physiol Regul Integr Comp Physiol. 1990;259:R1214–R1219. doi: 10.1152/ajpregu.1990.259.6.R1214. [DOI] [PubMed] [Google Scholar]
- 11.Chopard A, Pons F, Marini JF. Cytoskeletal protein contents before and after hindlimb suspension in a fast and slow rat skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2001;280:R323–R330. doi: 10.1152/ajpregu.2001.280.2.R323. [DOI] [PubMed] [Google Scholar]
- 12.Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 13.Cottin P, Poussard S, Mornet D, Brustis JJ, Mohammadpour M, Leger J, Ducastaing A. In vitro digestion of dystrophin by calcium-dependent proteases, calpains I and II. Biochimie. 1992;74:565–570. doi: 10.1016/0300-9084(92)90156-9. [DOI] [PubMed] [Google Scholar]
- 14.Cullen MJ, Walsh J, Nicholson LV, Harris JB, Zubrzycka-Gaarn EE, Ray PN, Worton RG. Immunogold labelling of dystrophin in human muscle, using an antibody to the last 17 amino acids of the C-terminus. Neuromuscul Disord. 1991;1:113–119. doi: 10.1016/0960-8966(91)90058-z. [DOI] [PubMed] [Google Scholar]
- 15.Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der MJ, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Camp-bell KP. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ervasti JM, Campbell KP. Dystrophin and the membrane skeleton. Curr Opin Cell Biol. 1993;5:82–87. doi: 10.1016/s0955-0674(05)80012-2. [DOI] [PubMed] [Google Scholar]
- 17.Faulkner JA, Brooks SV, Opiteck JA. Injury to skeletal muscle fibers during contractions: conditions of occurrence and prevention. Phys Ther. 1993;73:911–921. doi: 10.1093/ptj/73.12.911. [DOI] [PubMed] [Google Scholar]
- 18.Friden J, Lieber RL. Serum creatine kinase level is a poor predictor of muscle function after injury. Scand J Med Sci Sports. 2001;11:126–127. doi: 10.1034/j.1600-0838.2001.011002126.x. [DOI] [PubMed] [Google Scholar]
- 19.Hack AA, Ly CT, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamer PW, McGeachie JM, Davies MJ, Grounds MD. Evans Blue Dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J Anat. 2002;200:69–79. doi: 10.1046/j.0021-8782.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harlow E, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 1988. pp. 471–504. [Google Scholar]
- 22.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 23.Hori S, Ohtani S, Nguyen TM, Morris GE. The N-terminal half of dystrophin is protected from proteolysis in situ. Biochem Biophys Res Commun. 1995;209:1062–1067. doi: 10.1006/bbrc.1995.1605. [DOI] [PubMed] [Google Scholar]
- 24.Hunter KD, Faulkner JA. Pliometric contraction-induced injury of mouse skeletal muscle: effect of initial length. J Appl Physiol. 1997;82:278–283. doi: 10.1152/jappl.1997.82.1.278. [DOI] [PubMed] [Google Scholar]
- 25.Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- 26.Komulainen J, Takala TE, Kuipers H, Hesselink MK. The disruption of myofibre structures in rat skeletal muscle after forced lengthening contractions. Pflügers Arch. 1998;436:735–741. doi: 10.1007/s004240050696. [DOI] [PubMed] [Google Scholar]
- 27.Komulainen J, Takala TE, Vihko V. Does increased serum creatine kinase activity reflect exercise-induced muscle damage in rats? Int J Sports Med. 1995;16:150–154. doi: 10.1055/s-2007-972983. [DOI] [PubMed] [Google Scholar]
- 28.Lapointe BM, Cote CH. Anesthetics can alter subsequent in vitro assessment of contractility in slow and fast skeletal muscles of rat. Am J Physiol Regul Integr Comp Physiol. 1999;277:R917–R921. doi: 10.1152/ajpregu.1999.277.3.R917. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Mericskay M, Agbulut O, Butler-Browne G, Carlsson L, Thornell LE, Babinet C, Paulin D. Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J Cell Biol. 1997;139:129–144. doi: 10.1083/jcb.139.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lieber RL, Friden J. Muscle damage is not a function of muscle force but active muscle strain. J Appl Physiol. 1993;74:520–526. doi: 10.1152/jappl.1993.74.2.520. [DOI] [PubMed] [Google Scholar]
- 31.Lieber RL, Schmitz MC, Mishra DK, Friden J. Contractile and cellular remodeling in rabbit skeletal muscle after cyclic eccentric contractions. J Appl Physiol. 1994;77:1926–1934. doi: 10.1152/jappl.1994.77.4.1926. [DOI] [PubMed] [Google Scholar]
- 32.Lynch GS, Faulkner JA. Contraction-induced injury to single muscle fibers: velocity of stretch does not influence the force deficit. Am J Physiol Cell Physiol. 1998;275:C1548–C1554. doi: 10.1152/ajpcell.1998.275.6.C1548. [DOI] [PubMed] [Google Scholar]
- 33.Lynch GS, Rafael JA, Chamberlain JS, Faulkner JA. Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am J Physiol Cell Physiol. 2000;279:C1290–C1294. doi: 10.1152/ajpcell.2000.279.4.C1290. [DOI] [PubMed] [Google Scholar]
- 34.MacIntyre DL, Reid WD, Lyster DM, Szasz IJ, McKenzie DC. Presence of WBC, decreased strength, and delayed soreness in muscle after eccentric exercise. J Appl Physiol. 1996;80:1006–1013. doi: 10.1152/jappl.1996.80.3.1006. [DOI] [PubMed] [Google Scholar]
- 35.Malm C, Nyberg P, Engstrom M, Sjodin B, Lenkei R, Ekblom B, Lundberg I. Immunological changes in human skeletal muscle and blood after eccentric exercise and multiple biopsies. J Physiol. 2000;529:243–262. doi: 10.1111/j.1469-7793.2000.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem (Tokyo) 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- 37.McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan DL, Allen DG. Early events in stretch-induced muscle damage. J Appl Physiol. 1999;87:2007–2015. doi: 10.1152/jappl.1999.87.6.2007. [DOI] [PubMed] [Google Scholar]
- 39.Nosaka K, Clarkson PM. Relationship between post-exercise plasma CK elevation and muscle mass involved in the exercise. Int J Sports Med. 1992;13:471–475. doi: 10.1055/s-2007-1021300. [DOI] [PubMed] [Google Scholar]
- 40.Ohlendieck K. Towards an understanding of the dystrophin-glycoprotein complex: linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur J Cell Biol. 1996;69:1–10. [PubMed] [Google Scholar]
- 41.Patel TJ, Lieber RL. Force transmission in skeletal muscle: from actomyosin to external tendons. Exerc Sport Sci Rev. 1997;25:321–363. [PubMed] [Google Scholar]
- 42.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakuraba H, Hori S, Ohtani S, Hanaka S, Abe T, Shimmoto M, Suzuki Y. A case of Duchenne muscular dystrophy with truncated dystrophin. Significance of a cysteine-rich domain for functional expression of dystrophin protein. Brain Dev. 1993;15:222–225. doi: 10.1016/0387-7604(93)90069-k. [DOI] [PubMed] [Google Scholar]
- 44.Sam M, Shah S, Friden J, Milner DJ, Capetanaki Y, Lieber RL. Desmin knockout muscles generate lower stress and are less vulnerable to injury compared with wild-type muscles. Am J Physiol Cell Physiol. 2000;279:C1116–C1122. doi: 10.1152/ajpcell.2000.279.4.C1116. [DOI] [PubMed] [Google Scholar]
- 45.Spencer MJ, Croall DE, Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem. 1995;270:10909–10914. doi: 10.1074/jbc.270.18.10909. [DOI] [PubMed] [Google Scholar]
- 46.St Pierre SB, Brickson S, Corr DT, Best T. CD11b+ neutrophils predominate over RAM11+ macrophages in stretch-injured muscle. Muscle Nerve. 2002;25:837–844. doi: 10.1002/mus.10109. [DOI] [PubMed] [Google Scholar]
- 47.Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Street SF, Ramsey RW. Sarcolemma: transmitter of active tension in frog skeletal muscle. Science. 1965;149:1379–1380. doi: 10.1126/science.149.3690.1379. [DOI] [PubMed] [Google Scholar]
- 49.Thompson JL, Balog EM, Fitts RH, Riley DA. Five myofibrillar lesion types in eccentrically challenged, unloaded rat adductor longus muscle–a test model. Anat Rec. 1999;254:39–52. doi: 10.1002/(SICI)1097-0185(19990101)254:1<39::AID-AR6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 50.Tidball JG. Force transmission across muscle cell membranes. J Biomech. 1991;24:43–52. doi: 10.1016/0021-9290(91)90376-x. [DOI] [PubMed] [Google Scholar]
- 51.Tidball JG. Interactions between muscle and the immune system during modified musculoskeletal loading. Clin Orthop. 2002:S100–S109. doi: 10.1097/00003086-200210001-00012. [DOI] [PubMed] [Google Scholar]
- 52.Tidball JG, Berchenko E, Frenette J. Macrophage invasion does not contribute to muscle membrane injury during inflammation. J Leukoc Biol. 1999;65:492–498. [PubMed] [Google Scholar]
- 53.Tidball JG, Spencer MJ. Calpains and muscular dystrophies. Int J Biochem Cell Biol. 2000;32:1–5. doi: 10.1016/s1357-2725(99)00095-3. [DOI] [PubMed] [Google Scholar]
- 54.Warren GL, Hayes DA, Lowe DA, Armstrong RB. Mechanical factors in the initiation of eccentric contraction-induced injury in rat soleus muscle. J Physiol. 1993;464:457–475. doi: 10.1113/jphysiol.1993.sp019645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warren GL, Ingalls CP, Lowe DA, Armstrong RB. Excitation-contraction uncoupling: major role in contraction-induced muscle injury. Exerc Sport Sci Rev. 2001;29:82–87. doi: 10.1097/00003677-200104000-00008. [DOI] [PubMed] [Google Scholar]
- 56.Yoshida M, Suzuki A, Shimizu T, Ozawa E. Proteinase-sensitive sites on isolated rabbit dystrophin. J Biochem (Tokyo) 1992;112:433–439. doi: 10.1093/oxfordjournals.jbchem.a123918. [DOI] [PubMed] [Google Scholar]
- 57.Zerba E, Komorowski TE, Faulkner JA. Free radical injury to skeletal muscles of young, adult, and old mice. Am J Physiol Cell Physiol. 1990;258:C429–C435. doi: 10.1152/ajpcell.1990.258.3.C429. [DOI] [PubMed] [Google Scholar]