

Abstract
Peroxiredoxins make up a ubiquitous family of cysteine-dependent peroxidases that reduce hydroperoxide or peroxynitrite substrates through formation of a cysteine sulfenic acid (R-SOH) at the active site. In the 2-Cys peroxiredoxins, a second (resolving) cysteine reacts with the sulfenic acid to form a disulfide bond. For all peroxiredoxins, structural rearrangements in the vicinity of the active site cysteine(s) are necessary to allow disulfide bond formation and subsequent reductive recycling. In this study, we evaluated the rate constants for individual steps in the catalytic cycle of Salmonella typhimurium AhpC. Conserved Trp residues situated close to both peroxidatic and resolving cysteines in AhpC give rise to large changes in fluorescence during the catalytic cycle. For recycling, AhpF very efficiently reduces the AhpC disulfide, with a single discernible step and a rate constant of 2.3 × 107 M−1 s−1. Peroxide reduction was more complex and could be modeled as three steps, beginning with a reversible binding of H2O2 to the enzyme (k1 = 1.36 × 108 M−1 s−1, and k−1 = 53 s−1), followed by rapid sulfenic acid generation (620 s−1) and then rate-limiting disulfide bond formation (75 s−1). Using bulkier hydroperoxide substrates with higher Km values, we found that different efficiencies (kcat/Km) for turnover of AhpC with these substrates are primarily caused by their slower rates of binding. Our findings indicate that this bacterial peroxiredoxin exhibits rates for both reducing and oxidizing parts of the catalytic cycle that are among the fastest observed so far for this diverse family of enzymes.
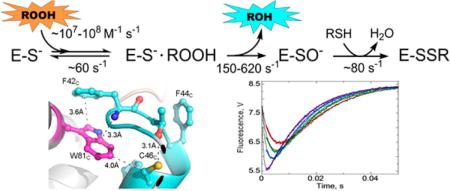
Peroxiredoxins (Prxs) are cysteine-dependent enzymes with major roles in peroxide defense and cell signaling in many, if not all, organisms, yet only a few studies have explored the pre-steady state kinetic profiles of these important enzymes.1–3 There are now numerous reports verifying peroxide reactivities as high as 107−108 M−1 s−1, where reactivity is defined either as kcat/Km for peroxide or as the second-order rate constant with H2O2 measured in competition experiments.3–7 How the highly conserved active site of Prxs serves to promote catalysis is an area of active investigation.8–10 Modulation of the active site Cys thiol pKa (to values of ~5–6) is an important factor in generating the reactive, nucleophilic thiolate at the active site,11,12 and the conserved Arg and Thr residues play important roles in this regard.13–15 Moreover, Arg and Thr in particular interact directly with the proximal oxygen atom of the incoming hydroperoxide substrate in ways that help stabilize the transition state and promote catalysis.8,9,16 As the structural and catalytic features around the active site become better defined, there remains a need for better information from rapid reaction kinetic approaches to more fully explore how these active sites so effectively recognize and turn over such simple, waterlike substrates.
In the reaction catalyzed by Prxs, the Cys thiolate attacks the hydroperoxide substrate, breaking the bond between the oxygens and forming Cys sulfenic acid (SOH) on the enzyme and the small molecule product (water or alcohol) in the active site (Figure 1A). This step is followed by a rearrangement, or local unfolding (LU), of residues around the peroxidatic Cys (denoted CP) to permit formation of a disulfide bond with another thiol, often contributed by another Cys within the Prx protein (designated the resolving Cys, or CR). Recycling of the disulfide through reductases like thioredoxin, glutaredoxin, or the AhpF flavoenzyme reactivates the Prx for another catalytic cycle. Structural and bioinformatic studies support the subdivision of Prx proteins into six subclasses that typically vary in the location of CR.14,17 The most widespread class, denoted the Prx1/AhpC or simply Prx1 class, has also been termed the typical 2-Cys Prx class,18,19 and its members possess a CR in the C-terminus that forms an intersubunit disulfide with CP, making these proteins obligate dimers (or multimers of dimers). These enzymes, which include bacterial AhpC and human PrxI–PrxIV, exhibit some of the fastest known peroxide reaction rates and are some of the best characterized in terms of steady state reaction kinetics.6,20,21
Figure 1.
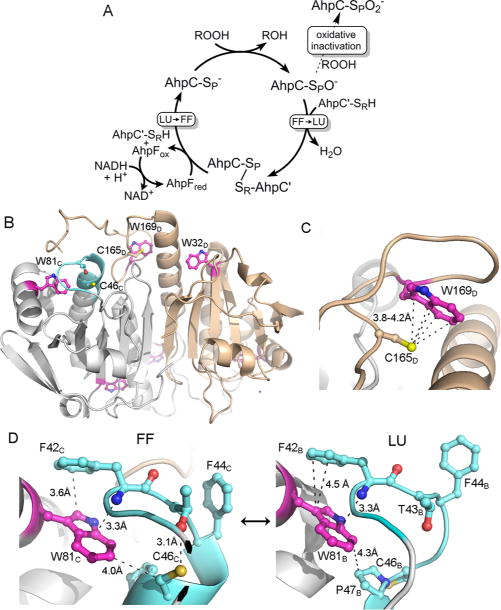
Catalytic cycle of 2-Cys peroxiredoxins and the location of the three Trp residues in Salmonella typhimurium AhpC. (A) The catalytic cycle (solid arrows) is shown along with the peroxidemediated inactivation pathway (dashed arrow). The conformation change necessary for Prx catalysis is shown as a transition from fully folded (FF) to locally unfolded (LU) enzyme after sulfenate formation at the peroxidatic Cys (shown as −SPO−), and the opposite transition that occurs upon reduction of the disulfide bond between SP and SR (the sulfur group of the resolving Cys) to regenerate the active enzyme. Oxidative inactivation by peroxide can also occur at the peroxidatic Cys (dashed arrow) to generate the cysteine sulfinate (shown as −SPO2−). (B) A single dimer [taken from a 1.8 Ắ resolution decameric crystal structure of reduced, wild-type AhpC (Protein Data Bank entry 4MA9)] is depicted with chain D colored tan and the partner subunit (chain C) colored gray, highlighting the location of Trp residues (magenta), Cys residues, and the regions that undergo structural changes (cyan) during catalysis (sulfur atoms colored yellow, nitrogen atoms blue, and oxygen atoms red). (C) Close-up of the region around C165 in the FF conformation. In the LU conformation, W169 is presumed to be disordered as no electron density is observed for it. (D) Close-ups of the region around W81 in the FF and LU conformations. Selected residues and atomic distances (dashed lines) in panels C and D are shown with the same color scheme as in panel B. Subscript letters after the residue numbers indicate the subunit of that residue (described in ref 30). Shown to the right in panel D is the LU conformation only of subunit B in Protein Data Bank entry 4MA9.
In this study, we set out to evaluate the rate constants for individual steps in the catalytic cycle of Salmonella typhimurium AhpC. Pre-steady state kinetics monitored by changes in fluorescence and absorbance features of AhpC were evaluated after addition of reductant, or of various hydroperoxide substrates, and compared with previously determined steady state parameters for this enzyme.21 For the disulfide recycling step, we observed a simple bimolecular electron transfer reaction between the reducing, CXXC-containing domain of AhpF and the oxidized, disulfide bond-containing AhpC at a rate that was nearly as fast as that of the oxidizing part of the cycle with peroxide. On the other hand, reduction of hydroperoxide substrates was more complex and was best modeled as three steps that we interpreted as reversible binding of the peroxide to the enzyme, followed by sulfenic acid formation on the enzyme (concomitant with water or alcohol generation) and then rate-limiting disulfide bond formation. This work also provided insights into how the reaction of AhpC with various hydroperoxides differentially affects the rates of the individual steps of the reaction.
EXPERIMENTAL PROCEDURES
Materials
Hydrogen peroxide (30%), dimethyl sulfoxide, and most buffer components were from Fisher. 1,4-Dithiothreitol (DTT) was obtained from Anatrace, Inc. (Maumee, OH). Cumene hydroperoxide and tert-butyl hydroperoxide were purchased from Sigma-Aldrich (St. Louis, MO). Ethyl hydroperoxide (5%) was purchased from Polysciences, Inc. (Warrington, PA).
Methods
Site-Directed Mutagenesis and Protein Preparation
The three Trp residues of StAhpC were substituted individually with Phe to generate the mutant proteins W32F, W81F, and W169F using the QuikChange protocol (Stratagene, Santa Clara, CA). Mutations introduced into the parent expression plasmid, pTHCm-ahpC11, were validated by sequencing the entire gene. Wild-type and mutant StAhpC were expressed in JW0598 (lacking ahpC) Escherichia coli cells22 grown in Studier’s ZYM-5052 autoinduction medium23 in the presence of 25 μg/mL chloramphenicol. The purification procedure for wild-type and mutant AhpC proteins was essentially the same as that described previously24. After purification by phenyl Sepharose and anion exchange chromatography, the proteins were concentrated by ultrafiltration, exchanged into 25 mM potassium phosphate (pH 7.0) and 1 mM EDTA and stored in aliquots at −80 °C until they were used. The concentrations of AhpC proteins were determined spectrophotometrically (extinction coefficients of 24300 M−1 cm−1 for wild-type AhpC at 280 nm and 17700–23100 M−1 cm−1 for Trp mutants, as summarized in Table S1 of the Supporting Information).
C165S and S128W NTD Expression and Purification
AhpC C165S was expressed and purified using the same procedure that was used for wild-type AhpC, except that the purification was conducted in the presence of a reducing agent (20 mM β-mercaptoethanol during column chromatography and dialysis, with the final storage buffer containing 2 mM DTT).
The S128W mutant of the N-terminal domain (NTD) fragment (residues 1–202) of AhpF and wild-type AhpF were expressed and purified as described previously; protein concentrations were assessed using an ɛ280 of 21250 M−1 cm−1 for S128W NTD6 and an ɛ450 of 13100 M−1 cm−1 for AhpF.24
Determination of Extinction Coefficients
Extinction coefficients for W32F, W81F, and W169F mutants were determined experimentally by comparisons of 280 nm absorbance and the protein content as measured by the microbiuret assay using bovine serum albumin as a standard25,26 (see Table S1 of the Supporting Information for extinction coefficients).
Standardization of Peroxide Solutions
The hydrogen peroxide concentration was determined spectrophotometrically (ɛ240 = 43.6 M−1 cm−1), and stock solutions of cumene hydroperoxide (initially diluted ~60-fold into dimethyl sulfoxide), tert-butyl hydroperoxide (t-butyl-OOH), and ethyl hydroperoxide were standardized on the basis of consumption of NADH (ɛ340 = 6220 M−1 cm−1) in an assay mixture containing AhpF, AhpC, and limiting peroxide, essentially as described previously (except with AhpF in place of thioredoxin and thioredoxin reductase).27
Preparation of Reduced Proteins
Wild-type and mutant StAhpC proteins as well as S128W NTD were incubated with a 20-fold excess of DTT for at least 1 h, followed by removal of DTT using a PD-10 desalting column (GE Healthcare) as described in detail elsewhere.27
Rapid Reaction Kinetic Studies
Proteins and peroxides were prepared in the same reaction buffer used previously for AhpF and AhpC steady state peroxidase assays [50 mM potassium phosphate (pH 7.0), 0.5 mM EDTA, and 100 mM ammonium sulfate], loaded into the syringes of an Applied Photophysics SX.18MV stopped-flow spectrophotometer, and allowed to equilibrate to 25 ± 0.1 °C. The absorbance or fluorescence changes were recorded after mixing (subject to a dead time of 1.5 ms); fluorescence was monitored at 90° relative to the incident light using excitation at 280 nm and emission at >320 nm (using a filter).
Data Analysis and Kinetic Simulations
Fits of the stopped-flow kinetic data were conducted using the Applied Photo-physics SX.18MV analysis software and single- or multiple-exponential fits as appropriate. Kinetic simulations made use of Gepasi, version 3.3,28 and KinTek Global Kinetic Explorer (Professional version 4.0.140728).29 The concentration series data with H2O2 or t-butyl-OOH were imported into KinTek Explorer and fit to a double-exponential equation with the aFit function to obtain an estimated standard deviation. Two kinetic models (with either two or three steps) were considered, and the rate constants and output factors were adjusted manually until a profile matching the data was reached. Initial estimates for rate constants were based on simple double-exponential fits and the concentration dependence of the two rate constants (shown in Figures 3A and 5B–D). For the more complex three-step model, one rate constant (k−1) was fixed while all others were allowed to float in the Data Fit Editor and the goodness of fit was determined by eye (“chi-by-eye”) and χ2/DoF (DoF, degrees of freedom). The FitSpace Editor was then employed to determine the confidence intervals for each of the fitted rate constants.
Figure 3.
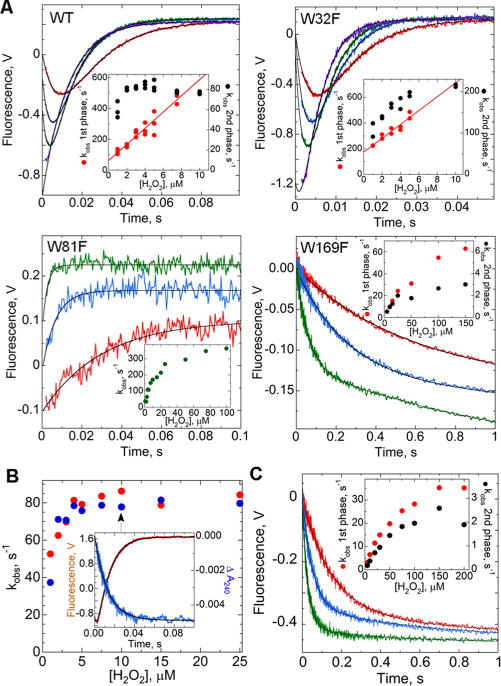
Kinetic profiles of intrinsic Trp fluorescence and absorbance changes of wild-type and mutant AhpC during H2O2-mediated oxidation. (A) Reduced AhpC (1 μM) in reaction buffer [25 mM potassium phosphate (pH 7) with 1 mM EDTA and 100 mM ammonium sulfate] at 25 °C was mixed in a stopped-flow spectrofluorometer with increasing concentrations of H2O2, with representative traces colored red, blue, green, and purple (1, 3, 5, and 20 μM for the wild type, 1, 3, 5, and 15 μM for W32F, 2, 10, and 100 μM for W81F, and 2, 4, and 25 μM for W169F). All concentrations are after mixing. All data (except for W81F) fit to a double exponential (black lines) yielded rate constants for the fast (red) and slow (black) phases, plotted vs H2O2 concentration in the insets. For ease of viewing, starting voltages for each set were adjusted to 0; note that voltages in each panel are not directly comparable. Traces for W81F, fit to a single exponential (with rate constants shown in the inset), have been offset consecutively by 0.1 V. (B) Same as panel A, with reduced AhpC (1 μM) mixed with a range of H2O2 concentrations, monitoring changes in absorbance at 240 nm (blue) and in fluorescence (red) in identical mixings. The slower rate constants from the fluorescence changes fit as in panel A match closely the rate constants for the absorbance changes at 240 nm (from a single-exponential fit). Shown in the inset are representative data from the experiment with 10 μM H2O2. (C) Similar to panel A, 2 μM reduced C165S AhpC was mixed with H2O2 at 10 (red), 50 (blue), and 200 μM (green).
Figure 5.
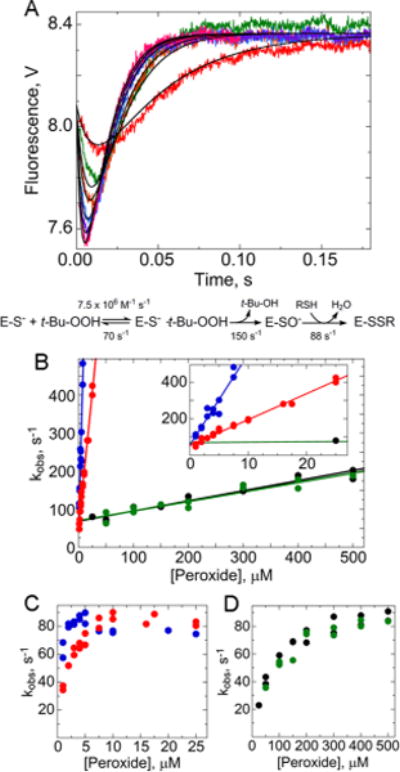
Rapid reaction kinetics of AhpC oxidation by various hydroperoxides. (A) Oxidation of wild-type AhpC by 50 (red), 150 (green), 200 (orange), 300 (blue), 400 (purple), and 500 μM tert-butyl hydroperoxide (t-Bu-OOH) (magenta) monitored by Trp fluorescence. Methods used were identical to those described in the legend of Figure 2. Data were fit to a three-step model as in Figure 4, shown below the plot. In panels B–D, blue indicates data for H2O2, red those for ethyl hydroperoxide, green those for t-Bu-OOH, and black those for cumene hydroperoxide. Results from two independent experiments are shown for each hydroperoxide. Panel B shows the rate of the fast, initial decrease for the four hydroperoxides, zooming in on values at low concentrations in the inset. Panels C and D show that all the substrates approach the same maximal rate for the exponential increase in fluorescence, with the slower substrates requiring higher concentrations.
RESULTS AND DISCUSSION
To evaluate the rate constants for individual steps in the catalytic cycle of S. typhimurium AhpC, we utilized pre-steady state assays set up to monitor partial turnover reactions. The rate of disulfide reduction in oxidized AhpC was determined by monitoring fluorescence changes associated with its reaction with the prereduced N-terminal domain (NTD) of AhpF. To monitor AhpC-catalyzed reduction of peroxide, we explored transient changes in intrinsic tryptophan fluorescence that occur as AhpC undergoes the transition from reduced to oxidized. This approach has been used previously to monitor pre-steady state kinetics of human PrxV and Mycobacterium tuberculosis AhpE.1,2 Fluorescence spectra and lifetimes of the more closely related chloroplast 2-Cys Prx from barley also supported the potential utility of measuring Trp fluorescence during AhpC oxidation.20 AhpC contains three tryptophan residues (Figure 1B–D), two of which are highly conserved within the Prx1 subfamily [Trp81 and Trp169 (see Conservation of AhpC Trp Residues in the Supporting Information and ref 20)]. While Trp32 is distant from the active site, Trp81 packs against the Cβ atom of Cys46 (CP) in reduced AhpC, where it could potentially sense local conformational changes around the CP thiolate during substrate binding and catalysis (Figure 1B,D). Structural comparison of the FF and LU states of AhpC indicates that the local environment surrounding Trp81 changes during this transition (Figure 1D). This is also true for Trp169, which packs closely against Cys165 (CR) (Figure 1C) when in the FF conformation but exhibits no electron density in any of the LU AhpC structures.30,31
Kinetics of Electron Transfer between AhpF and AhpC
Beginning with the CP–CR disulfide form of AhpC, a dedicated flavoprotein reductase known as AhpF catalyzes the generation of dithiol AhpC ready for reacting with peroxide (Figure 1A). The interactions between AhpF and AhpC have been extensively studied with regard to the steady state kinetics of their interaction32–34 and the identity and role of each of the catalytic Cys residues in these proteins.32,35–37 A CXXC motif in the N-terminal domain (NTD) of AhpF is the direct donor of electrons to AhpC, ultimately deriving the reducing power from NADH via the other two redox centers (FAD and another CXXC) found in the thioredoxin reductase-like, C-terminal domains of AhpF.38 For use in stopped-flow studies, we have shown that an independently expressed NTD with a Ser128Trp mutation (S128W NTD) exhibits no significant differences in kinetic behavior as an electron donor to AhpC yet shows substantial differences in fluorescence between the oxidized and reduced forms, allowing accurate tracking of reaction kinetics.6,39
To assess the pre-steady state kinetics of the reduction of AhpC by AhpF, prereduced S128W NTD at various concentrations (in excess) was mixed with oxidized AhpC and the total Trp fluorescence of both proteins was monitored (Figure 2A). As determined from the slope of the line in Figure 2B, electron transfer between the two proteins was a second-order process with a rate constant of 2.3 × 107 M−1 s−1. This value matches well with the kcat/Km for S128W NTD of 1.0 × 107 M−1 s−1 that was measured previously by steady state assays.21 This part of the catalytic cycle is therefore a very efficient process for supporting peroxide reduction in this enzyme system.
Figure 2.
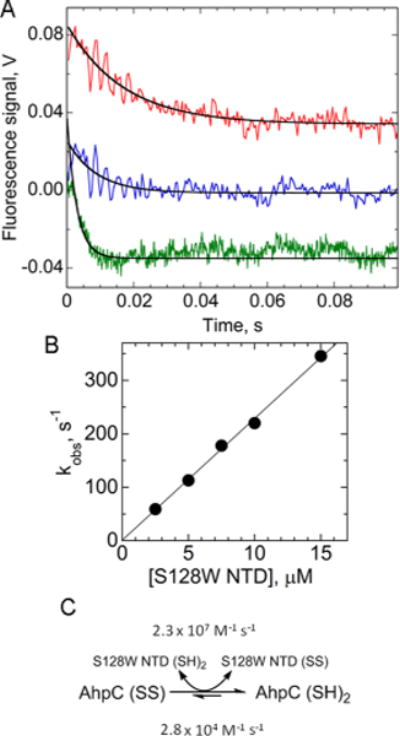
Reduction of AhpC by the redox-sensing mutant of the AhpF N-terminal domain (S128W NTD). (A) To assess rates of reduction of AhpC, oxidized AhpC (1 μM for 2.5–10 μM S128W NTD and 3 μM for >10 μM S128W NTD) was mixed with reduced S128W NTD at 2.5 (red), 5 (blue), and 15 μM (green), monitoring fluorescence changes in both proteins over time (λex = 280 nm; λem > 320 nm). Red and blue curves have been displaced upward on the y-axis for the sake of clarity; amplitudes of change were somewhat variable in these averaged, noisy spectra, but the data were readily fit to a single-exponential equation. (B) Pseudo-first-order rate constants obtained from the fits in panel A, plotted vs S128W NTD concentration, yield a second-order rate constant of 2.3 × 107 M−1 s−1. (C) Reaction pathway annotated with rate constants.
Using the known redox potentials at pH 7 for the dithiol and disulfide forms of both the NTD (E°′ = −264 mV)36 and AhpC (E°′ = −178 mV),21 the Nernst equation [ΔE = (RT)/(nF) ln Keq] yields an equilibrium constant of 811:1 in favor of oxidized NTD and reduced AhpC at a 1:1 ratio of the two proteins. Given the forward rate constant for electron transfer reported above, the reverse rate constant of the electron transfer (from reduced AhpC to oxidized NTD) can be calculated as 2.8 × 104 M−1 s−1 (Figure 2C).
Comparing these findings regarding the reduction cycle with those from other Prxs and their reductases, we note that the rate of Prx reduction has been directly measured, using the same approach as reported here, for human PrxV with human mitochondrial thioredoxin (Trx2), and M. tuberculosis Tpx with M. tuberculosis TrxB, giving rates of 2 × 106 M−1 s−1 and 8.2 × 103 M−1 s−1, respectively.1,2 The apparent second-order rate constant for reduction of additional Prxs has been calculated from steady state measurements, and these range from 2 × 104 to 1 × 107 M−1 s−1, as summarized by Trujillo et al.40 Thus, reduction of AhpC by the NTD of AhpF constitutes one of the most efficient Prx recycling reactions reported, highlighting one catalytic advantage gained by expression of a specialized AhpC reductase in these bacteria.
Pre-Steady State Kinetics of AhpC Reacting with H2O2
Although the steady state fluorescence emission spectra are rather similar for both oxidized and reduced AhpC,11 an analysis of the millisecond-scale fluorescence changes at low H2O2 concentrations readily demonstrated two phases of fluorescence changes; the fluorescence initially decreases for 2–10 ms and then increases over the next 50–70 ms to a value slightly above the starting fluorescence (Figure 3A), indicating that changes in Trp fluorescence are detecting at least two distinct kinetic steps occurring during the oxidation of AhpC by peroxide. The magnitude of these fluorescence changes is quite large; beginning with the reduced enzyme, the fluorescence decreases by up to 30% (e.g., upon addition of 10 μM H2O2) and then increases again to a final value ~5% above the starting fluorescence. Under the partial turnover conditions used for these experiments, the two thiols in each active site of AhpC are oxidized to a disulfide bond; we do not observe any peroxide-dependent sulfinic acid formation in the resistant wild-type AhpC.41,42
The fluorescence changes observed reflect alterations in the environments of some combination of the three Trp residues of AhpC (Figure 1B–D). To help evaluate the contribution of each Trp residue to the observed fluorescence changes, individual Trp mutants of AhpC were prepared and evaluated for their time-dependent fluorescence changes after addition of H2O2. The largest loss of fluorescence emission intensity is observed when Trp32 is replaced with Phe (W32F) (Figure S1 of the Supporting Information), but this mutant, which retains the highly conserved Trp81 and Trp169 of Prx1 family members, exhibits the same pattern of fluorescence changes upon addition of H2O2 as the wild-type enzyme does (a decrease and then an increase), which in this case occurs approximately twice as fast (complete within ~30 ms). This is consistent with the ~3-fold higher kcat and the ~1.3-fold higher kcat/Km for H2O2 observed in steady state analyses of this mutant (Table S2 of the Supporting Information). These data suggest that the pre-steady state changes in fluorescence of the wild-type enzyme are not substantially influenced by Trp32.
The effects of replacing either of the other two Trp residues with Phe on the time-dependent fluorescence changes in the stopped-flow experiments are much greater; for W81F, the fluorescence rises only a very small amount upon addition of H2O2, whereas for W169F, it only decreases (Figure 2A). We infer from the altered fluorescence profiles of these mutants that Trp81 [which is near CP (Figure 1D)] is primarily responsible for the initial decrease in fluorescence after mixing H2O2 with the wild-type enzyme, and that Trp169 [near CR (Figure 1C)] is primarily responsible for the subsequent increase in fluorescence.
We next analyzed the rates of the fluorescence changes observed after addition of H2O2 to AhpC. For wild-type AhpC, when data are fit to a biphasic (double-exponential) process, the rate constant (kobs) obtained for the initial fluorescence decrease is linearly dependent on the H2O2 concentration (Figure 3A, wild type); this is even true when equimolar substrate (1 μM) is added. The non-zero intercept of this line also implies that this phase is reversible. Note that at H2O2 concentrations of >15 μM, the first phase is so rapid that it is essentially complete within the 1.5 ms dead time of the stopped-flow instrument [rate constants of >500 s−1 (Figure 3A)]. Unlike that for the first phase, the rate constant for the second phase plateaus with the increasing H2O2 concentration (black circles in Figure 3A) and reaches a limiting value by ~2 (wild type) or ~10 μM H2O2 (W32F). The decrease in fluorescence observed for W169F (Figure 3A) fits best to a triple-exponential model, but only the first rate is fast enough to be catalytically competent for this enzyme [which has a kcat of ~17 s−1 (Table S2 of the Supporting Information)]. In fact, W169F is the most catalytically impaired enzyme, with a decreased ~100-fold compared with that of wild-type AhpC (Table S2 of the Supporting Information).
Distinct from the other two mutants and wild-type AhpC, W81F exhibits fluorescence changes during reaction with H2O2 that are best fit by a single exponential. It is notable that the rate constant obtained, which should reflect the second phase of the reaction sensed primarily by Trp169, is considerably faster than that of the second phase observed for wild-type and W32F proteins. It therefore appears that either the second phase is occurring more quickly in this mutant or the first phase more slowly, such that the two become difficult to distinguish during curve fitting. Confirmation that the reaction of H2O2 with W81F is slower than with wild-type AhpC was obtained using a H2O2 competition assay and horseradish peroxidase as the competing enzyme (method described in ref 11). In these studies, W81F was found to react with H2O2 at approximately half the rate of the wild-type enzyme at pH 7 (Table S3 of the Supporting Information).
The slopes of the lines defined by the observed rate constants for the first phase plotted against H2O2 concentration for wild-type and W32F AhpC yield second-order rate constants of 5.0 × 107 and 5.4 × 107 M−1 s−1, respectively, for the forward reaction and associated y-intercepts (first-order rate constants) of 56 and 174 s−1, respectively (average of three experiments each). This second-order rate constant for the forward reaction is quite similar to the kcat/KmHOOH value obtained by previous steady state measurements of AhpC (3.7 × 107 M−1 s−1)21 and with the second-order rate constant measured using competition assays with horseradish peroxidase (3.2 × 107 M−1 s−1).11 In part on the basis of the location of the Trp residues predominantly responsible for each phase, we hypothesized that the first, peroxide-dependent phase reflects the association with peroxide, where the slope provides the rate constant for peroxide binding (k1) and the intercept provides the rate of dissociation (k−1); the peroxide-independent, limiting second rate constant then represents either the reaction between the enzyme and the substrate or subsequent disulfide bond formation within the enzyme.
In an effort to further clarify the nature of the H2O2-mediated fluorescence changes in AhpC, we monitored changes in absorbance at 240 nm after H2O2 addition, which could reflect changes in the thiolate content of the enzyme. Monitoring of the 240 nm absorbance of wild-type AhpC across a range of pH values was previously shown to be useful in determining the pKa of CP.11 Over a range of H2O2 concentrations from 1 to 25 μM, A240 decreased by ~0.005 (ɛ240 of ~5000 M−1 cm−1) with a first-order rate constant that closely matched the rate obtained for the second phase (increase) in fluorescence observed for AhpC (Figure 3B). These data would be consistent with the second phase of the reaction corresponding to one (or more) steps that lead to the loss of thiolate (i.e., the step involves chemistry). However, the meaning of the changes in absorbance at 240 nm is not unambiguous. In particular, we have no knowledge of the spectral features of sulfenic acid or sulfenate in this region of the UV spectrum, but studies with several small molecules stabilizing sulfenic acids have previously reported absorbance features in this region of the spectrum.43,44 Furthermore, with the significant conformational changes associated with disulfide bond formation in AhpC, the environment of various residues is shifted and could contribute to the observed absorbance changes at 240 nm.
To help in assessing the contribution of disulfide bond formation to the pattern of fluorescence changes observed for AhpC, the C165S mutant that lacks CR was studied (Figure 3C). This mutant cannot form the disulfide bond but, unlike wild-type AhpC, does ultimately react with a second H2O2 to become hyperoxidized to the sulfinic acid. Also, unlike for oxidized wild-type AhpC that is locked into its LU disulfide form, the relative amounts of oxidized C165S in the FF and LU conformations are unknown. Fluorescence changes after addition of H2O2 to reduced C165S AhpC led to a decrease in fluorescence, but no increase, consistent with the increase in fluorescence of the wild-type enzyme being caused by the trapping of the wild-type enzyme in the LU, disulfide-bonded form. Despite the lack of a subsequent increase in fluorescence, the decrease in C165S fluorescence still fits best to a double-exponential equation (inset of Figure 3C), with both of the rate constants reflecting rather slow reactions. This suggests that C165S is significantly inhibited in substrate binding and/or catalysis compared to wild-type AhpC. Competition assays evaluating the reactivity of C165S AhpC toward H2O2 relative to horseradish peroxidase support this hypothesis as C165S was found to exhibit ~14% of the reactivity of the wild-type enzyme at pH 7 (Table S3 of the Supporting Information).
Kinetic Model for the Reaction of H2O2 with AhpC
To identify a reaction mechanism that would be consistent with our experimental data, we fit the kinetic data to two-step (A + B ↔ C → D) and three-step (A + B ↔ C → D → E) models using KinTek Explorer. The difference between the two models is the representation of the reactions involved in sulfenic acid and disulfide bond formation with these being combined into a single step in the simpler model (C → D) and shown as discrete states in the more complex model (C → D → E). This approach showed a much better fit to the data for the three-step model (χ2/DoF = 3.32) than for the two-step model (χ2/DoF = 11.4) (Figure 4 and Figure S2 of the Supporting Information, also evident using “chi-by-eye”). The three-step model includes four rate constants (k1, k−1, k2, and k3) and scaling factors for the fluorescence of four species (reduced, H2O2-bound, sulfenic acid, and disulfide AhpC forms). Through multiple iterations, we found that holding the least well determined rate constant, k−1, fixed at 53 s−1 (based on the y-intercept of the linear fit of the first phase using data from 1 to 7.5 μM H2O2), we could fit on all seven other parameters to give well-constrained rate constants (Figure S3 and Table S4A of the Supporting Information). Values for k1, k2, and k3 after fitting to this model (Figure 4) were 1.36 × 108 M−1 s−1, 622 s−1, and 74.5 s−1, respectively (Table S4A of the Supporting Information). As shown in the model (Figure 4), we interpret these steps as binding of H2O2, chemical reaction to form sulfenic acid on the enzyme, and rate-limiting disulfide bond formation, respectively. These assignments are consistent with our results with C165S, which exhibits no fluorescence increase and also no disulfide bond formation (Figure 3C). On the basis of these results, we infer that the A240 changes occurring coincident with the second phase of the reaction (Figure 3B) reflect the disappearance of sulfenic acid as the disulfide bond is formed. We note that the value of k1 obtained by this approach is ~4-fold higher than was previously obtained for H2O2 reaction by competition kinetics11 and by the simple interpretation of the slope of kobs versus H2O2 concentration for the initial fluorescence decrease (Figure 3A); we expect that this new value more accurately represents the true reactivity of StAhpC with H2O2 as it takes into account the sequential steps of the reaction and includes a second kinetic step that is not readily recognized using the simpler kinetic analysis. Moreover, these stopped-flow kinetic studies with multiple substrate concentrations combined with global fitting using KinTek Explorer are likely to be quantitatively more reliable than the competition assays which use single, substoichiometric amounts of H2O2.11
Figure 4.
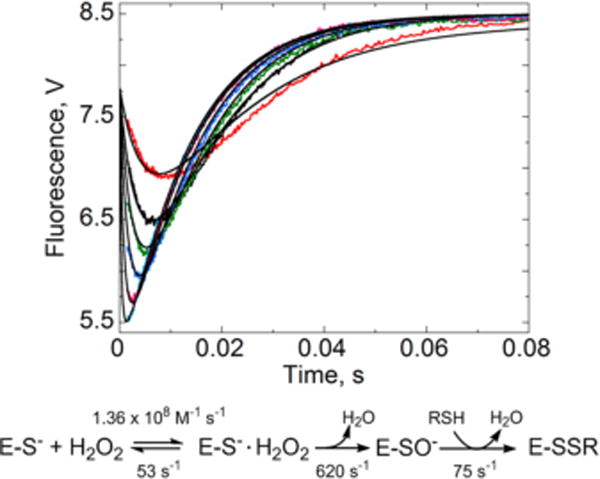
Kinetic model for fitting AhpC fluorescence changes after addition of H2O2. KinTek Global Kinetic Explorer was used to test kinetic models and conduct fits to the data as described in the text. The best fits to the global model are the black lines shown with the experimental data (gathered as in Figure 3a) at various H2O2 concentrations, colored red (1 μM), black (2 μM), green (3 μM), blue (5 μM), magenta (10), and cyan (25 μM). The kinetic model to which these data fit best and our interpretation of the reaction steps detected are depicted below the plot.
Kinetic Profiles of AhpC with Peroxides Other Than H2O2
In addition to H2O2, bulkier hydroperoxides are also substrates of AhpC, albeit with higher Km values (but the same kcat values).21 Considering alternatives to the tiniest substrate, H2O2, one would expect binding to be highly affected by the nature of the substrate, but not k3, which we argue reflects intramolecular disulfide bond formation. The chemical reaction between the hydroperoxide and CP, once the substrate is bound, might be affected by the nature of the substrate. We therefore repeated the experiments in Figure 3 with the other substrates previously studied by steady state assays.21
Using these bulkier hydroperoxides as substrates, the pattern of the fluorescence changes was quite similar to what was seen with H2O2, but higher ROOH concentrations were required to elicit them in a manner consistent with the increased Km values for these substrates (Figure 5). Particularly notable from these experiments are the very similar values for the y-intercept of the linear fits of the first phase of the double-exponential curve for each of these substrates (values from 53.8 to 70.4 s−1) as well as the limiting rate of the second phase (from 81 to 86 s−1) (Figure 5B–D and Table S5 of the Supporting Information). As seen in Figure 5B, it is the slope of kobs versus ROOH concentration for the first phase of fluorescence changes that is highly affected by the nature of the substrate, indicating that some combination of changes in binding (k1) and peroxide reduction (k2) is slowed for these poorer substrates. To better understand these effects, the data using the poorest substrate, t-butyl-OOH, were fit to the same three-step model used in the fits of the H2O2 data. If the rate for k−1 (at 70.4 s−1) based on the y-intercept of kobs versus the concentration of t-butyl-OOH (Figure 5B) is fixed as we did when modeling the H2O2 data of Figure 3A, the most significant change in rate constant after fitting the data with the three-step model was for k1, which was decreased ~180-fold for t-butyl-OOH compared with that for H2O2 (7.5 × 105 M−1 s−1) (Table S4C of the Supporting Information). The value for k2 was also decreased, but to a much smaller degree (by ~4-fold, to 150 s−1). Thus, the lower catalytic efficiencies exhibited by the larger substrates like t-butyl-OOH and cumene hydroperoxide reflect the first phase of the reaction, and particularly the rate of binding of the substrate to the enzyme.
Perspective
Kinetic data collected using fluorescence and absorbance changes associated with catalysis by StAhpC, as well as simulations to fit the data to a sequential three-step mechanism, are consistent with a model in which the peroxide substrate binds and then reacts with CP, followed by rate-limiting disulfide bond formation between CP and CR. Not included in the model, but arguably important for catalysis and the pattern of fluorescence changes observed, is the possibility that the binding of the peroxide induces movement of amino acid side chains proximal to Trp81, leading to enhanced quenching of its fluorescence. Thus, we suspect that the fluorescence changes are not due to a direct effect of the noncovalent binding of H2O2, but rather to its effect on the conformation of the active site. Additional complexity in the binding of peroxide substrates is also suggested by other aspects of our findings. The data presented imply that the Kd for binding of H2O2 is ~400 nM, a value that seems quite low given that this very small molecule can form, at most, only six hydrogen bonds (with two H-bond donors and four electron pairs to act as H-bond acceptors). Furthermore, the changes in binding and catalysis using bulkier substrates are observed primarily as decreases in k1, rather than increased off rates (k−1), and this conceptually makes more sense if k1 includes an “induced fit” step that is not as efficient for the bulkier substrates. While we cannot yet claim that our data prove such a conformational change step before catalysis, these arguments are at least suggestive that such a change occurs.
With this work, StAhpC joins a small group of Prxs for which rates of some of the individual steps of the reaction have been determined. To date, only one other Prx, from the narrowly distributed AhpE subfamily, has exhibited a rate constant with its own best substrate (lipid hydroperoxide) with a similar magnitude, at 1.8 × 108 M−1 s−1, as the initial step of peroxide binding in StAhpC (1.3 × 108 M−1 s−1).2 (Note that the previously reported second-order rate constant for human PrxII reacting with H2O2, which was also in the range of 108 M−1 s−1, was determined by competition kinetics.5) A member of a third subfamily studied by rapid reaction kinetic approaches, human PrxV, exhibited rates of 106–107 M−1 s−1 for the first step of reaction with its preferred substrates, peroxynitrite and organic hydroperoxides.1 For PrxV and AhpE, H2O2 was a relatively poor substrate, giving rates of ~105 M−1 s−1 for the first step.1,2
Intramolecular disulfide bond formation rates are also now known for several different Prxs. The fastest so far is StAhpC, at 75 s−1, while human PrxIII and PrxV exhibit intermediate values of 22 and 15 s−1, respectively.1,45 The slowest disulfide bond formation was observed for human PrxII, at 1.7 s−1, and it is no coincidence that this enzyme is also highly susceptible to inactivation during turnover with peroxides. As demonstrated elegantly by Peskin et al.,45 the difference in hyperoxidation sensitivity between PrxII and PrxIII tracks directly with their rate of disulfide bond formation; the sulfenic acid form of PrxII persists longer and has a greater chance to become hyperoxidized before a disulfide bond can form. Fitting with this perspective, StAhpC, with a relatively high disulfide bond formation rate of 75 s−1, is quite insensitive to inactivation during turnover.
In summary, we found through these rapid reaction kinetic studies that AhpF reduces the disulfide of AhpC very efficiently and in a single step with a forward rate constant of 2.3 × 107 M−1 s−1 and a reverse rate constant of 2.8 × 104 M−1 s−1 (Figure 2). Peroxide reduction was more complex and could be modeled as three steps, beginning with a reversible binding of H2O2 to the enzyme (k1 = 1.36 × 108 M−1 s−1, and k−1 = 53 s−1), followed by rapid sulfenic acid generation (620 s−1) and then rate-limiting disulfide bond formation (74.5 s−1) (Figures 3A and 4). The rate-limiting step, at ~80 s−1 for all hydroperoxides, is consistent with an overall kcat of ~52 s−1 obtained from steady state measurements.21 In addition, these studies show that different efficiencies (kcat/Km) for turnover of AhpC with bulkier hydroperoxide substrates are primarily caused by their slower rates of binding.
Supplementary Material
Acknowledgments
Funding
This study was supported by a grant from the National Institutes of Health to L.B.P. and P.A.K. (RO1 GM050389). Partial support is also acknowledged from an Interim Bridge Funding Award from Wake Forest School of Medicine (L.B.P.) and a Summer Honors Research Fellowship from Emory & Henry College Friends of the Sciences (S.A.).
ABBREVIATIONS
- Prx
peroxiredoxin
- StAhpC
S. typhimurium alkyl hydroperoxide reductase C component (peroxidase)
- AhpF
alkyl hydroperoxide reductase F component (flavoprotein reductase)
- CP
peroxidatic cysteine
- CR
resolving cysteine
- DTT
1,4-dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- S128W NTD
mutant of the truncated form of AhpF containing only residues 1-202 at the N-terminus, with Ser128 mutated to Trp
- t-butyl-OOH
tert-butyl hydroperoxide
Footnotes
Supporting Information
Additional results, including extinction coefficients, fluorescence properties and steady state assay results with the Trp mutants, HRP competition assays comparing mutant and wildtype StAhpC proteins, summaries of the results obtained after fitting of the time-dependent fluorescence data to two- and three-step kinetic models, and bioinformatics information about the conservation of each of the Trp residues in StAhpC. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Trujillo M, Clippe A, Manta B, Ferrer-Sueta G, Smeets A, Declercq JP, Knoops B, Radi R. Pre-steady state kinetic characterization of human peroxiredoxin 5: Taking advantage of Trp84 fluorescence increase upon oxidation. Arch Biochem Biophys. 2007;467:95–106. doi: 10.1016/j.abb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 2.Trujillo M, Mauri P, Benazzi L, Comini M, De Palma A, Flohe L, Radi R, Stehr M, Singh M, Ursini F, Jaeger T. The mycobacterial thioredoxin peroxidase can act as a one-cysteine peroxiredoxin. J Biol Chem. 2006;281:20555–20566. doi: 10.1074/jbc.M601008200. [DOI] [PubMed] [Google Scholar]
- 3.Hugo M, Turell L, Manta B, Botti H, Monteiro G, Netto LE, Alvarez B, Radi R, Trujillo M. Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: Kinetics, acidity constants, and conformational dynamics. Biochemistry. 2009;48:9416–9426. doi: 10.1021/bi901221s. [DOI] [PubMed] [Google Scholar]
- 4.Horta BB, de Oliveira MA, Discola KF, Cussiol JR, Netto LE. Structural and biochemical characterization of peroxiredoxin Qβ from Xylella fastidiosa: Catalytic mechanism and high reactivity. J Biol Chem. 2010;285:16051–16065. doi: 10.1074/jbc.M109.094839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manta B, Hugo M, Ortiz C, Ferrer-Sueta G, Trujillo M, Denicola A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch Biochem Biophys. 2009;484:146–154. doi: 10.1016/j.abb.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 6.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 8.Hall A, Parsonage D, Poole LB, Karplus PA. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J Mol Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrer-Sueta G, Manta B, Botti H, Radi R, Trujillo M, Denicola A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol. 2011;24:434–450. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 10.Zeida A, Reyes AM, Lebrero MC, Radi R, Trujillo M, Estrin DA. The extraordinary catalytic ability of peroxiredoxins: A combined experimental and QM/MM study on the fast thiol oxidation step. Chem Commun. 2014;50:10070–10073. doi: 10.1039/c4cc02899f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson KJ, Parsonage D, Hall A, Karplus PA, Poole LB. Cysteine pKa values for the bacterial peroxiredoxin AhpC. Biochemistry. 2008;47:12860–12868. doi: 10.1021/bi801718d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poole LB, Hall A, Nelson KJ. Current Protocols in Toxicology. Wiley; New York: 2011. Overview of peroxiredoxins in oxidant defense and redox regulation; pp. 7.9.1–7.9.15. Chapter 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagy P, Karton A, Betz A, Peskin AV, Pace P, O’Reilly RJ, Hampton MB, Radom L, Winterbourn CC. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: A kinetic and computational study. J Biol Chem. 2011;286:18048–18055. doi: 10.1074/jbc.M111.232355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall A, Nelson K, Poole LB, Karplus PA. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid Redox Signaling. 2011;15:795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salsbury FR, Jr, Yuan Y, Knaggs MH, Poole LB, Fetrow JS. Structural and electrostatic asymmetry at the active site in typical and atypical peroxiredoxin dimers. J Phys Chem B. 2012;116:6832–6843. doi: 10.1021/jp212606k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portillo-Ledesma S, Sardi F, Manta B, Tourn MV, Clippe A, Knoops B, Alvarez B, Coitino EL, Ferrer-Sueta G. Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry. 2014;53:6113–6125. doi: 10.1021/bi500389m. [DOI] [PubMed] [Google Scholar]
- 17.Nelson KJ, Knutson ST, Soito L, Klomsiri C, Poole LB, Fetrow JS. Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins. 2011;79:947–964. doi: 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: Alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood ZA, Schroder E, Harris JR, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 20.König J, Lotte K, Plessow R, Brockhinke A, Baier M, Dietz KJ. Reaction mechanism of plant 2-Cys peroxiredoxin. Role of the C terminus and the quaternary structure. J Biol Chem. 2003;278:24409–24420. doi: 10.1074/jbc.M301145200. [DOI] [PubMed] [Google Scholar]
- 21.Parsonage D, Karplus PA, Poole LB. Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc Natl Acad Sci USA. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Poole LB, Ellis HR. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 25.Bailey JL. Techniques in Protein Chemistry. Elsevier Scientific Publishing Co., Inc.; New York: 1967. [Google Scholar]
- 26.Janatova J, Fuller JK, Hunter MJ. The heterogeneity of bovine albumin with respect to sulfhydryl and dimer content. J Biol Chem. 1968;243:3612–3622. [PubMed] [Google Scholar]
- 27.Nelson KJ, Parsonage D. Current Protocols in Toxicology. Wiley; New York: 2011. Measurement of peroxiredoxin activity; pp. 7.10.11–7.10.28. Chapter 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendes P. GEPASI: A software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput Appl Biosci. 1993;9:563–571. doi: 10.1093/bioinformatics/9.5.563. [DOI] [PubMed] [Google Scholar]
- 29.Johnson KA, Simpson ZB, Blom T. Global kinetic explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 30.Perkins A, Nelson KJ, Williams JR, Parsonage D, Poole LB, Karplus PA. The sensitive balance between the fully folded and locally unfolded conformations of a model peroxiredoxin. Biochemistry. 2013;52:8708–8721. doi: 10.1021/bi4011573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: Redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 32.Jönsson TJ, Ellis HR, Poole LB. Cysteine reactivity and thiol-disulfide interchange pathways in AhpF and AhpC of the bacterial alkyl hydroperoxide reductase system. Biochemistry. 2007;46:5709–5721. doi: 10.1021/bi7001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niimura Y, Poole LB, Massey V. Amphibacillus xylanus NADH oxidase and Salmonella typhimurium alkyl-hydroperoxide reductase flavoprotein components show extremely high scavenging activity for both alkyl hydroperoxide and hydrogen peroxide in the presence of S. typhimurium alkyl-hydroperoxide reductase 22-kDa protein component. J Biol Chem. 1995;270:25645–25650. doi: 10.1074/jbc.270.43.25645. [DOI] [PubMed] [Google Scholar]
- 34.Poole LB, Higuchi M, Shimada M, Calzi ML, Kamio Y. Streptococcus mutans H2O2-forming NADH oxidase is an alkyl hydroperoxide reductase protein. Free Radical Biol Med. 2000;28:108–120. doi: 10.1016/s0891-5849(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 35.Poole LB, Reynolds CM, Wood ZA, Karplus PA, Ellis HR, Li Calzi M. AhpF and other NADH:peroxiredoxin oxidoreductases, homologues of low Mr thioredoxin reductase. Eur J Biochem. 2000;267:6126–6133. doi: 10.1046/j.1432-1327.2000.01704.x. [DOI] [PubMed] [Google Scholar]
- 36.Reynolds CM, Poole LB. Attachment of the N-terminal domain of Salmonella typhimurium AhpF to Escherichia coli thioredoxin reductase confers AhpC reductase activity but does not affect thioredoxin reductase activity. Biochemistry. 2000;39:8859–8869. doi: 10.1021/bi000826d. [DOI] [PubMed] [Google Scholar]
- 37.Reynolds CM, Poole LB. Activity of one of two engineered heterodimers of AhpF, the NADH:peroxiredoxin oxidoreductase from Salmonella typhimurium, reveals intrasubunit electron transfer between domains. Biochemistry. 2001;40:3912–3919. doi: 10.1021/bi002766h. [DOI] [PubMed] [Google Scholar]
- 38.Wood ZA, Poole LB, Karplus PA. Structure of intact AhpF reveals a mirrored thioredoxin-like active site and implies large domain rotations during catalysis. Biochemistry. 2001;40:3900–3911. doi: 10.1021/bi002765p. [DOI] [PubMed] [Google Scholar]
- 39.Parsonage D, Reeves SA, Karplus PA, Poole LB. Engineering of fluorescent reporters into redox domains to monitor electron transfers. Methods Enzymol. 2010;474:1–21. doi: 10.1016/S0076-6879(10)74001-5. [DOI] [PubMed] [Google Scholar]
- 40.Trujillo M, Ferrer-Sueta G, Thomson L, Flohe L, Radi R. Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Subcell Biochem. 2007;44:83–113. doi: 10.1007/978-1-4020-6051-9_5. [DOI] [PubMed] [Google Scholar]
- 41.Nelson KJ, Parsonage D, Karplus PA, Poole LB. Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Methods Enzymol. 2013;527:21–40. doi: 10.1016/B978-0-12-405882-8.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 43.Heckel A, Pfleiderer W. Lumazinesulfenates: A new class of stable sulfenic acids. Tetrahedron Lett. 1983;24:5047–5050. [Google Scholar]
- 44.Tripolt R, Belaj F, Nachbaur E. Unexpectedly stable sulfenic acid: 4,6-Dimethoxy-l,3,5-triazine-2-sulfenic acid; synthesis, properties, molecular and crystal structure. Z Naturforsch. 1993;48b:1212–1222. [Google Scholar]
- 45.Peskin AV, Dickerhof N, Poynton RA, Paton LN, Pace PE, Hampton MB, Winterbourn CC. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem. 2013;288:14170–14177. doi: 10.1074/jbc.M113.460881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.