

Abstract
This study aimed to evaluate genetic variability in the FUS and TDP-43 genes, known to be mainly associated with amyotrophic lateral sclerosis (ALS), in patients with the diagnoses of frontotemporal lobar degeneration (FTLD) and corticobasal syndrome (CBS). We screened the DNA of 228 patients for all the exons and flanking introns of FUS and TDP-43 genes. We identified 2 novel heterozygous missense mutations in FUS: P106L (g.22508384>T) in a patient with behavioral variant frontotemporal dementia (bvFTD) and Q179H in several members of a family with behavioral variant FTD. We also identified the N267S mutation in TDP-43 in a CBS patient, previously only reported in 1 ALS family and 1 FTD patient. Additionally, we identified 2 previously reported heterozygous insertion and deletion mutations in Exon 5 of FUS; Gly174-Gly175 del GG (g. 4180–4185 delGAGGTG) in an FTD patient and Gly175-Gly176 ins GG (g. 4185–4186 insGAGGTG) in a patient with diagnosis of CBS. Not least, we have found a series of variants in FUS also in neurologically normal controls. In summary, we report that genetic variability in FUS and TDP-43 encompasses a wide range of phenotypes (including ALS, FTD, and CBS) and that there is substantial genetic variability in FUS gene in neurologically normal controls.
Keywords: Frontotemporal dementia, Corticobasal syndrome, Genetics, FUS, TDP-43
1. Introduction
In 2009, in 2 separate studies, fused in sarcoma/translated in liposarcoma (FUS/TLS) gene was identified to be involved in pathogenesis of amyotrophic lateral sclerosis (ALS) (Kwiatkowski et al., 2009; Vance et al., 2009). FUS is a gene located on chromosome 16p11.2, encoding a 526 amino acid (aa) long protein which binds to RNA and DNA and regulates DNA repair, transcription regulation, RNA splicing, and cellular localization (Vance et al., 2009). TAR (trans-activation response)-DNA binding protein 43 (TDP-43) colocalizes with ubiquitinated protein deposits in the brain of frontotemporal dementia (FTD) and ALS patients (Neumann et al., 2006). TDP-43 gene mutations have been identified in a subset of ALS cases (Kabashi et al., 2008; Sreedharan et al., 2008). While both TDP-43 and FUS mutations have been associated with ALS, screening of frontotemporal lobar degeneration (FTLD) without motor neuron disease (MND) has rarely been associated with FUS or TDP-43 mutations. To our knowledge, only 1 behavioral variant FTD (bvFTD) patient has been associated with a TDP-43 mutation (Borroni et al., 2009) and 1 with a FUS mutation (Van Langenhove et al., 2010).
As we believed that similar pathology in FTD and ALS cases with TDP-43 positive deposits could imply to similar genetic defects, we sequenced the coding regions and flanking introns of FUS and TDP-43 genes to genetic variability in FUS and TDP-43 variants in our cohort of 158 FTLD, 70 corticobasal syndrome (CBS) patients and 21 members of an autosomal dominant inherited behavioral variant FTD family (see details below).
2. Methods
2.1. Study population
Blood samples from 228 index patients with the diagnosis of FTD (n = 158) or CBS (n = 70) were collected at the Cognitive Neuroscience Division of National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health (NIH). This cohort included patients with a diagnosis of bvFTD, progressive nonfluent aphasia (PNFA), semantic dementia (SD) (Neary et al., 1998), and CBS (Boeve, 2011). Five FTD patients had concomitant motor neuron disease. Subjects were seen as part of an ongoing research study on FTD and CBS at the Cognitive Neuroscience Section of the NINDS of the NIH, Bethesda, MD, USA. They were either self-referred or referred by outside neurologists. Patients arrived at the NIH with a caregiver and were diagnosed based on an initial clinical evaluation and examination by a neurologist by standard clinical criteria (Boeve, 2005; Neary et al., 1998). They then spent 9 days participating in extensive neuropsychological and neurologic testing and imaging studies. Their diagnoses were re-evaluated by a neuropsychologist (JG) based on the results of the testing performed at the NIH. We required all subjects to have an assigned research durable power of attorney prior to admission to the protocol and the assigned individuals gave written informed consent for the study. The patients gave assent for the study. All aspects of the study and the consent procedure were approved by the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute on Aging (NIA) Institutional Review Boards.
In this study we also included a family with FTD with an apparent autosomal dominant mode of inheritance from South Africa with Dutch ancestry (Afrikaner) (Fig. 1A). This family was collected through a collaborative effort between the Institute for Ageing and Health at Newcastle University and the University of Stellenbosch. We screened 21 members of the family, of which 8 were affected. We had previously reported a stop mutation in CHMP2B in 2 unaffected individuals of the family (Momeni et al., 2006). The members of this family had consented prior to blood draw according to the ethics committee of the University of Newcastle. The DNA was extracted from blood at the University of Newcastle using the standard procedures.
Fig. 1.
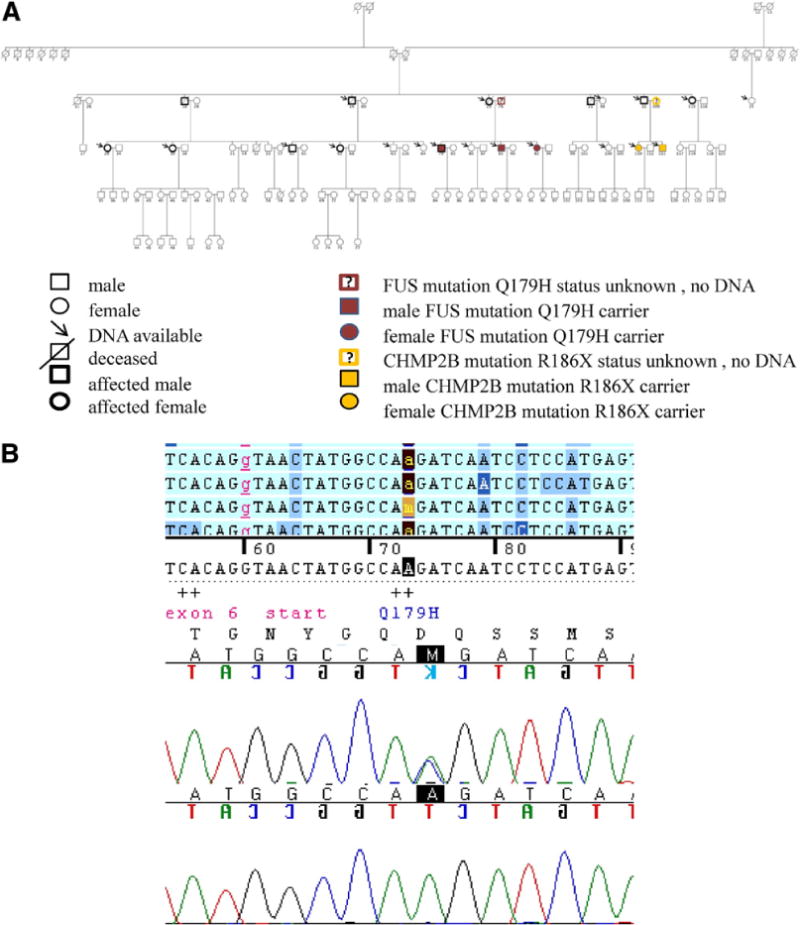
(A) Afrikaner family pedigree. The DNA from the individuals marked with an arrow was available for the genetic screening. Indivulas 79 (affected) and 81 and 82 (unaffected) carry the FUS Q179H variant but their affected mother and another unaffected sibling (individual 80) did not carry the variant. DNA from their father (individual 78) was not available for screening. (B) The electropherogram of heterozygous Q179H CAA>CAC variant, in FUS exon 6, individual 79 (the upper sequence) and the wild type sequence in individual 80 (the lower sequence).
2.2. Genetic analysis
Genomic DNA was extracted from peripheral blood using standard procedures. All 15 exons of FUS and all 6 exons of TDP-43 were sequenced as previously described (Sreedharan et al., 2008; Vance et al., 2009). All these individuals had been previously screened for mutations in MAPT, PGRN, and CHMP2B. The sequencing reactions were run on ABI 3730XL gene analyzer (ABI, Fosters City, CA, USA). The sequences were analyzed using Sequencher 4.9 software (Gene Codes Corporation, Ann Arbor, MI, USA). To determine the prevalence of the variants found in patients, we screened 569 normal controls from Coriell Institute for FUS variants and 174 neurologically normal controls (plates NDTP 096 and NDTP 098 from Coriell Institute) for the TDP-43 mutation N267S.
3. Results
Demographic data of the patients in which we identified variants/mutations are summarized in Table 1. Sequencing analysis of FUS in 228 patients with the diagnosis of FTD and CBS identified 2 missense variants, 1 deletion and 1 insertion, 2 synonymous changes, and 1 variant in the 3′ untranslated region (3′-UTR) (Table 2). One novel variant P106L in exon 4 was identified in an FTD patient (FTD 182; Fig. 2A and B). A heterozygous 6-base pair deletion in exon 5 Gly174-Gly175 del GG (g. 4180–4185 delGAGGTG) was identified in an FTD patient (FTD 85; Fig. 3B). This mutation had been previously reported in a familial amyotrophic lateral sclerosis (FALS) case (Kwiatkowski et al., 2009) but this variant was isolated in 2/638 controls in another study (Van Langenhove et al., 2010). Hence, presence of this variant in normal controls (Van Langenhove et al., 2010) suggests that this mutation is probably nonpathogenic. A heterozygous insertion in exon 5 Gly175-Gly176 ins GG (g. 4185–4186 insGAGGTG) was observed in a patient with the diagnosis of CBS (CBS 135; Fig. 3C). This variant had been previously reported by Kwiatkowski et al. (2009) in an ALS case but we found this variant in 1/659 of our normal controls. There were no family members of the index patients available to examine the segregation of the above-mentioned mutations, however, the existence of this variant among the neurologically normal controls most likely excludes the possibility of its involvement in the pathogenesis of these diseases. We also found N267S mutation in TDP-43 gene in a CBS patient (Fig. 4A and B). This mutation had been previously reported in an ALS patient (Corrado et al., 2009) and in an FTD patient without motor neuron disease (Borroni et al., 2009). We did not find this mutation in 174 neurologically normal controls.
Table 1.
Mean (standard deviation) demographic and clinical characteristics of 158 FTLD patients and 70 CBS patients. Maximum score on the MDRS2 is 144 points
| M/F | Mean age of onset (y) | Mean age at testing (y) | Mean education (y) | MDRS2 total score | R/L handed | FTLD subtype | |
|---|---|---|---|---|---|---|---|
| FTLD patients | 84/74 | 55.8 (8.5) | 60.1 (8.3) | 15.7 (2.8) | 101.0 (31.2) | 138/19/1 ambidextrous | 122 bv-FTD, 25 PPA, 5 FTD-MND, 6 SD |
| CBS patients | 51/37 | 60.1 (8.2) | 65.2 (7.7) | 14.7 (2.6) | 111.9 (23.3) | 81/6/1 ambidextrous | — |
Key: bv, behavioral variant; CBS, corticobasal syndrome; F, female; FTD, ; FTLD, frontotemporal lobar degeneration; L, left; M, male; MDRS2, ; MND, ; PPA, ; R, right; SD, semantic dementia.
Table 2.
Genetic variants in the FUS and TDP-43 genes identified among the FTD and CBS patients
| Patient number | Diagnosis | Mutation | Exon | Previously reported | Present in controls |
|---|---|---|---|---|---|
| FUS | |||||
| 182 | FTD | P106L CCC>CTC | 4 | No | No |
| 85 | FTD | Gly174-Gly175 del GG (g. 4180–4185 delGAGGTG) | 5 | FALS (Kwiatkowski et al., 2009) NC (Van Langenhove et al., 2010) |
No |
| 135 | CBS | Gly175-Gly176 ins GG (g. 4185–4186 insGAGGTG) | 5 | ALS (Kwiatkowski et al., 2009) | Yes (1/659) |
| 593-1 | FTD | P125P CCC>CCG | 5 | No | |
| AF-79 | FTD | Q179H CAA>CAC | 6 | No | |
| AF-81 | NC | Q179H CAA>CAC | 6 | No | |
| AF-82 | NC | Q179H CAA>CAC | 6 | No | |
| 568-1 | FTD | R522R AGG>AGA | 15 | Yes (1/659) | |
| 87 | CBS | 3′ UTR exon 15 STOP +41 | 3′UTR | Yes (9/659) | |
| 109 | CBS | 3′ UTR exon 15 STOP +41 | 3′UTR | Yes (9/659) | |
| TDP-43 | |||||
| 125 | CBS | N267S | ALS (Corrado et al., 2009); FTLD (Borroni et al., 2009) | Not tested |
Key: ALS, amyotrophic lateral sclerosis; CBS, corticobasal syndrome; FALS, familial amyotrophic lateral sclerosis; FTD, ; FTLD, frontotemporal lobar degeneration; NC, normal control; UTR, untranslated region.
Fig. 2.
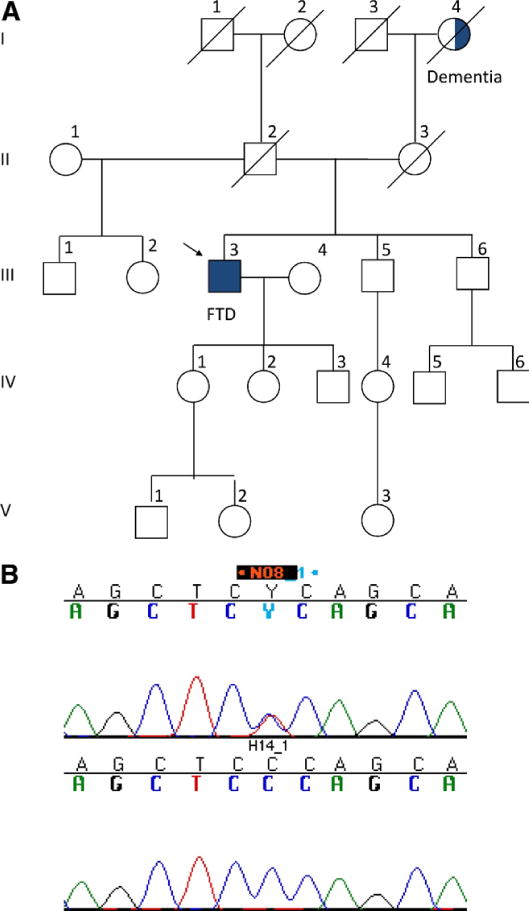
(A) The pedigree of patient with frontotemporal dementia FTD 182. The index patient is marked with an arrow. (B) The electropherogram of the variant P106L CCC>CTC in exon 4 of FUS gene in the upper panel. The lower panel shows a wild type sequence of the same region in a normal control DNA.
Fig. 3.
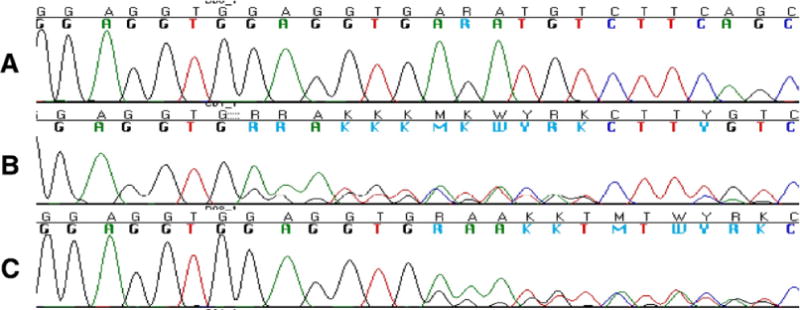
Wild type, deletion, and insertion in FUS exon 5. (A) The electropherogram of wild type sequence in FUS Exon 5. (B) The electropherogram of Gly174-Gly175 del GG (g. 4180–4185 delGAGGTG) variant in patient with frontotemporal dementia FTD 85. (C) The electropherogram of a heterozygous insertion in exon 5 Gly175-Gly176 insGG (g. 4185–4186insGAGGTG) in patient with corticobasal syndrome CBS135.
Fig. 4.
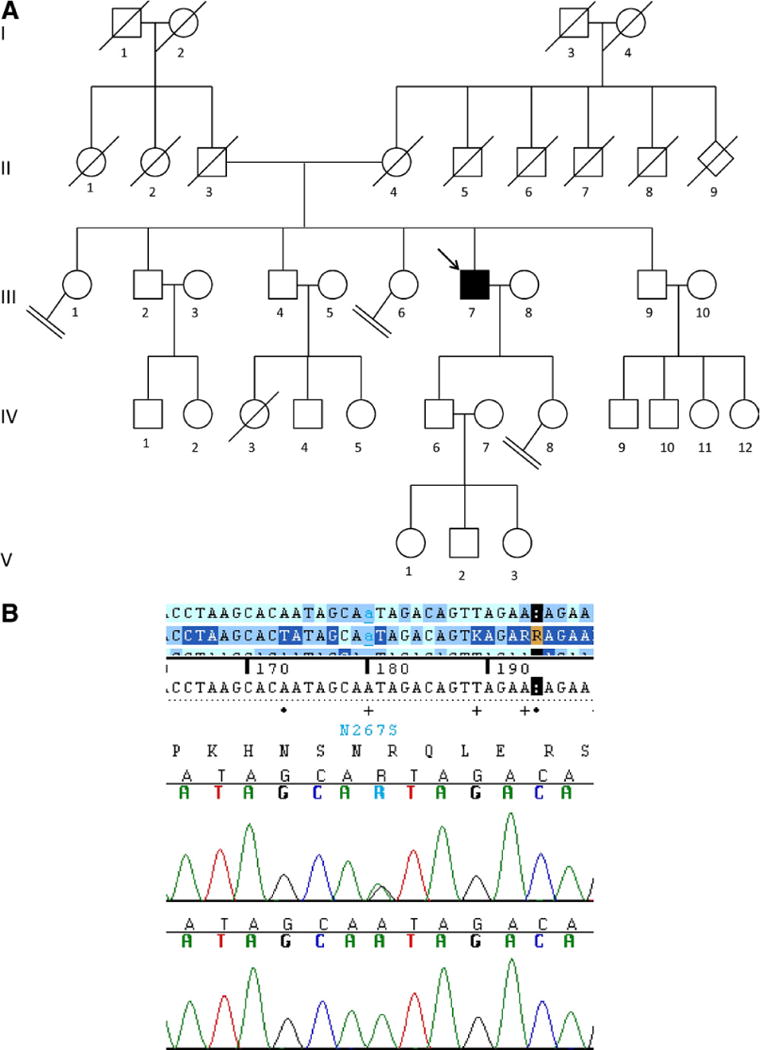
Patient with corticobasal syndrome CBS 125. (A) Pedigree of patient CBS125. The index patient is marked with an arrow. (B) The electropherogram showing the mutation exon 6 N267S in TDP-43. The upper electropherogram shows the mutated sequence in CBS 125 and the lower shows the wild type sequence.
Screening of the Afrikaner family with autosomal dominant bvFTD revealed Q179H variant in FUS exon 5 in only 1 branch of the family (affected individual 79, unaffected individuals 81 and 82 in pedigree; Fig. 1A and B).
To verify the prevalence of any of these variants among the neurologically normal controls, we screened FUS exons 4, 5, 6, and 15 in 659 DNA samples of normal controls from Coriell Institute. The overall results of our findings in patients are summarized in Table 2. A detailed list of variants in the normal controls with the DNA ID of Coriell Institute can be found in Supplementary Table 1. Below we describe the cases of missense mutation found exclusively in patients.
3.2. Case reports
3.2.1. Afrikaner family
This is a multigenerational pedigree with an apparent autosomal dominant bvFTD (Fig. 1). The clinical phenotype in this family is marked by executive dysfunction, social disinhibition, and dementia with the age of onset in the late 40s and early 50s. Findings of brain pathology are available for 3 affected siblings (numbers 19, 20, and 22 in the pedigree) with widespread tauopathy involving the gray and white matter. The DNA of 21 members of the family has been available for genetic screening. The DNA samples were screened for mutations in MAPT, PGRN, CHMP2B, FUS, and TDP-43. We have previously reported a CHMP2B nonsense mutation in 2 asymptomatic members of the family which has supposedly been inherited from the asymptomatic mother and not the affected father who did not carry the mutation (Momeni et al., 2006). Screening of FUS gene in affected members of the family revealed the Q179H mutation in exon 6 in 1 affected individual (number 79) but not the other affected family members. To understand the origin of the variant, we screened the DNA from the affected mother and the siblings. The mother who died of FTD at the age of 74 years did not carry the variant but 2 of the 3 siblings who are still asymptomatic carry the variant. The DNA from the father was not available to be tested for segregation but his medical history shows that he was not affected by any neurological disorder. As neither the mother nor any other affected member of the family carried the Q179H variant, the unaffected father is the obligate carrier of the variant. Although we did not identify this variant in any of the neurologically normal controls, and there is no proof of the cosegregation of the variant with the disease, we assume that this is a rare nonpathogenic variant. To identify the origin and the prevalence of this variant in Afrikaner population, further cases and normal controls will need to be collected and screened from the same population.
3.2.2. FTD 182
This patient was evaluated approximately 5 years ago at NIH (pedigree; Fig. 2A). At the time he was a 50-year-old right-handed man. Five years prior to his evaluation, he had developed symptoms of personality and behavior changes and executive dysfunction. He began buying multiple items he did not need. He developed apathy. His wife reported that his personality changed and that “he is not the same man I married.” He performed socially inappropriate behaviors such as telling childish jokes in inappropriate situations. He had difficulty remembering recent events and got lost in familiar situations. He had decreased language production. He had no insight into his symptoms. Three years after the start of his illness, he had to retire from his job because of his cognitive and behavior symptoms. His maternal grandmother had a history of dementia, but at a later age (Fig. 2A). His mother died prior to the mean age of onset of FTLD. On his neurological examination, he had hypometric saccades and glabellar, snout, and grasp reflexes. His affect was inappropriate. He perseverated and repeated responses. He had normal strength and no muscle atrophy or fasciculations.
Neuropsychological evaluation revealed diffuse deficits across nearly all aspects of cognition apart from basic attention and speed. Specifically, relative strengths included immediate auditory attention in the low average range as measured with the Digit Span subtest of the Wechsler Adult Intelligence Scales Third Edition (WAIS-3), visual attention in the mildly impaired range as measured with Wechsler Adult Intelligence Scales Third Edition Picture Completion, and motor speed in the mildly impaired range on the Trail Making subtest of the Delis-Kaplan Executive Function System (D-KEFS) (Wechsler 1987, 1997; Kaplan et al., 1983). All higher level verbal and nonverbal cognitive abilities, measured both with a global screening (the Mattis Dementia Rating Scale-2) as well as more specific tests of memory, language, and executive abilities, were moderately to severely impaired (Mattis 1976).
The patient had a magnetic resonance image (MRI) of the brain which showed diffuse atrophy. He had an FDG-PET (fluorodeoxyglucose-positron emission tomography) scan which showed moderate reductions in cerebral glucose metabolism greatest in the parietal-temporal cortex, but also in the frontal cortex of the left hemisphere with less reduction of glucose metabolism in the right hemisphere.
He met criteria for, and was given a diagnosis of, behavioral variant FTD with some language symptoms noted as well (Neary et al., 1998). His symptom presentation and age of onset are within the range observed in behavioral variant FTD. His relatively preserved attention and speed, and the parietal-temporal hypometabolism on FDG-PET are less typical. Since the patient’s evaluation at NIH, his symptoms have gradually worsened. He has become increasingly apathetic, his language production has deteriorated, and he is incontinent of urine and feces at times.
3.2.3. CBS 125
This patient was seen approximately 5 years ago at NIH (pedigree; Fig. 4). At the time, he was a 78-year-old man. Two years before he was seen at NIH, he began having poor balance with falls. He then developed left arm dystonia, clumsiness, and tremor. He had visuospatial symptoms with difficulty getting lost and navigating his way around furniture. He reported some difficulty remembering people’s names. He had some apathy and mild agitation, and no problems with swallowing. On neurological examination, he demonstrated proximal and distal transitive and intransitive apraxia, worse on the left, left visual hemineglect, poor smooth pursuit eye movements, left-sided sensory deficits and impaired stereognosis, and increased tone in his left arm and leg. He had alien limb findings on the left. Gait was unsteady and apraxic, with decreased left arm swing. On neuropsychological testing, he showed severe impairment on visuospatial tasks, moderately impaired nonverbal memory and language function, and executive dysfunction. Magnetic resonance imaging revealed moderate cortical and caudate atrophy. An FDG-PET scan showed reduced glucose metabolism in frontal, temporal, and parietal cortexes of both hemispheres, but with greater involvement of the right hemisphere. He met clinical criteria for CBS with motor and cognitive symptoms (Boeve, 2005). At this time, he has not come to autopsy.
The clinical information of patients FTD 85 and CBS 135 can be found in the Supplementary data. The electropherogram of the variants Gly174-Gly175 del GG (g. 4180–4185 delGAGGTG) variant in patient FTD 85 and the heterozygous insertion in exon 5 Gly175-Gly176 insGG (g. 4185–4186insGAGGTG) in patient CBS 135 are depicted in Fig. 3.
4. Discussion
FUS (fused in sarcoma) genetic variability has been reported to be linked to ALS (Kwiatkowski et al., 2009; Vance et al., 2009). TDP-43 and FUS are DNA-RNA binding proteins and TDP-43 is a major component of ubiquitinated protein aggregates ALS and FTLD. Wild type TDP-43 and FUS are mainly localized in the nucleus but ALS patients carrying FUS mutations do not show TDP-43 aggregates (Vance et al., 2009). Hyperphosphorylated TDP-43 has been found in the cortical neurons of patients with FTLD and spinal cord neurons of ALS patients (Neumann et al., 2006). As mutations in the genes encoding components of the protein deposits in the brain have been found to be causal in cases such as tauopathies, a number of studies were undertaken to identify genetic variants in TDP-43 gene in a spectrum of cases with FTD, FTD-ALS, and ALS. To date, pathogenic variants in TDP-43 have been more often linked to ALS and FTLD with motor neuron disease than to FTLD with behavioral and language variants, although TDP-43 has been found in the majority of frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) brains (Benajiba et al., 2009; Borroni et al., 2009; Gijselinck et al., 2009; Kovacs et al., 2009; Rollinson et al., 2007; Schumacher et al., 2009). In ALS cases, there are 29 pathogenic variants identified in TDP-43 (Kabashi et al., 2008; Rutherford et al., 2008; Daoud et al., 2009; Alzheimer Disease & Frontotemporal Dementia Mutation Database [www.molgen.ua.ac.be/ADMutations]). In this study, we screened a cohort of FTD (n = 158) and CBS (n = 70) patients counting 5 cases of FTLD with overlapping symptoms of FTLD and ALS or the co-occurrence of FTD and ALS in the same generation of the family, plus 21 members (8 of which affected) of a family with apparent autosomal dominant FTD. Screening of TDP-43 gene resulted in the identification of 1 pathogenic variant (Borroni et al., 2009; Corrado et al., 2009) in a patient with corticobasal syndrome and 6 nonpathogenic variants. We did not find this mutation in 174 neurologically normal controls. The screening of the FUS gene in this cohort led to the identification of 2 missense variants, 1 deletion and 1 insertion, 2 synonymous changes, and 1 variant in the 3′ UTR.
We identified the missense variant P106L in exon 4 of FUS in 1 patient with behavioral variant FTD. This is a novel variant and the first reported in exon 4. The amino acid 106 lies in the QGSY (Gln-Gly-Ser-Tyr) region of the FUS protein. Whether or not this variant cosegregates with the disease could not be verified due to lack of DNA samples from informative members of the family (Fig. 2A). However, this variant was not found in 659 neurologically normal controls. The pathogenicity of this variant will need to be verified either through the screening of other patients or functional analysis.
The variant Q179H in FUS exon 5 was identified in individuals 79, 81, and 82 of the Afrikaner pedigree (Fig. 1A and B). This variant is also located in the QSYG (Gln-Ser-Tyr-Gly) domain of FUS protein. As the pedigree structure in this family shows, an autosomal dominant mode of inheritance could be anticipated. The clinical manifestations of the disease and the age of onset are similar in all the affected individuals. The mother’s (number 20) brain and 2 of her siblings revealed profound frontotemporal lobar degeneration and neurofibrillary pathology characterized by widespread hyperphosphorylated tau inclusions in glial cells within the white matter (Kalaria et al., in preparation). All the family members, for which the DNA has been available, have been screened for MAPT, PGRN, CHMP2B, and TDP-43. In 2006 we reported a mutation in CHMP2B in 2 unaffected individuals of this family but not in their affected father. This case has been extensively discussed in Momeni et al. (2006). The FUS variant was isolated in 3 out of 4 siblings (Fig. 1A). The mother of the siblings had FTLD but did not carry the mutation. There is no DNA available from the father to be screened, but he is the obligate carrier of this variant. In an isolated case, this variant would have been assumed to be pathogenic. In this family however, the availability of the DNA from the unaffected siblings and other members of the extended pedigree made it possible to assess the inheritance and the possible role of this variant. Individual number 79, who was diagnosed 4 years ago at the age of 56, has shown a similar course of disease as the other family members. The siblings, individuals 81 and 82 (Fig. 1A), who are marginally younger in age have not shown any clinical symptoms. Additionally, as we did not detect the mutation in the affected mother or any other affected or unaffected family member and because the unaffected father is the obligate carrier of this variant, it is highly unlikely that this variant in FUS gene is pathogenic or that the family has a heterogenic genetic cause for FTD.
As seen in Table 2 and Supplementary Table 1, we have identified additional variants in the patients that we did not discuss further including Gly174-Gly175 del GG (g. 4180–4185 delGAGGTG) in a patient with diagnosis of FTLD (the detailed clinical information can be found in the Supplementary data). Nevertheless, it is noteworthy that this variant has been first reported in an ALS patient (Kwiatkowski et al., 2009) but in a later report it was identified in 2/638 normal controls aged 70 and 37 years, hence suggested to be nonpathogenic (Van Langenhove et al., 2010). We also isolated the Gly175-Gly176 ins GG (g. 4185–4186 insGAGGTG) variant in a patient with the diagnosis of CBS (the detailed clinical information can be found in Supplementary data). This variant had been previously reported by Kwiatkowski et al. (2009) in an ALS patient. We found this variant in 1/659 neurologically normal controls. This is the first report of this variant in a normal control suggesting that this variant could be nonpathogenic. The synonymous variant P125P was identified in an FTLD patient and none of our controls. Another synonymous variant R522R was found in 1 FTLD patient and 1 control. One variant in the 3′ UTR region of FUS 41 base pairs after stop codon was found in 2 patients and 9 controls. We found the other missense variants, S135N rs61732970, G227D, and the insertion deletions GGC.GGC.GGC.GGC.GGT to GGC.GGT Gly228_Gly230del (Belzil et al., 2009), (GGC) 5/6/7 Gly224 (rs72550890) 11 or 9 Gly, and GGA ins R244, only in our controls (see Supplementary Table 1).
Although variants in FUS have been reported to be causative of ALS, it is difficult to argue through what mechanism the same variants would be involved in a protective mechanism in neurologically normal controls. As we did not have access to the family members of the patient FTD 182, we cannot prove the cosegregation of the variant P106L with the disease. However, this variant was not present in our 659 neurologically normal controls. Further screening will shed light on the frequency and, perhaps, pathogenicity or nonpathogenicity of this variant/mutation.
In the Afrikaner familial case, the carriers of the variant Q179H are related to the extended FTD pedigree through their mother, who did not carry that variant (Fig. 1), suggesting that this variant is most likely nonpathogenic.
Previous studies that identified FUS/TLS mutations in ALS patients demonstrated that the ALS patients did not show cognitive deficits (Kwiatkowski et al., 2009; Vance et al., 2009). None of the patients in this study (see Table 1 and Supplementary Table 1b) had signs or symptoms of ALS. ALS and FTLD can have overlapping clinical and pathological features. Clinically, behavioral, cognitive, and language dysfunctions can be seen in patients primarily diagnosed with ALS and, pathologically, common areas of the brain show degeneration: the posterior frontal lobe atrophy in FTD-MND cases, or motor neurons and corticospinal tract in the cases of DHDL (dementia lacking distinctive histopathology), or the precentral gyrus, substantia nigra, and amygdala together with subcortical gliosis and neuronal loss in anterior cyngulate gyrus in ALS-FTD cases (Ferrari et al., 2011b). There is also overlap at the molecular pathology level with ubiquitin and TDP-43 positive inclusions in both FTLD and ALS (Ferrari et al., 2011b). Further, a region on chromosome 9 appears to be associated with both FTLD and ALS (Ferrari et al., 2011b), and there is a possible pathogenic effect of genetic variability in the valosin-containing protein gene (VCP) in ALS and FTD-ALS (Ferrari et al., 2011a).
CBS has a distinct clinical and pathological presentation from ALS. Given that our findings suggest that the variability in FUS gene is greater in the general population than previously assumed (Kwiatkowski et al., 2009; Van Langenhove et al., 2010), examination of more patients with the diagnoses of FTLD, ALS, and FTLD-ALS, and related disorders may be warranted.
The patient CBS 125 who carries the mutation N267S in exon 6 of TDP-43 is discussed in detail in the case report section. This mutation had been previously reported in 1 ALS and 1 FTD patient (Borroni et al., 2009; Corrado et al., 2009). The patient CBS 125 is the first report of this mutation in a case with the diagnosis of CBS.
To summarize, in our study we screened 228 cases of patients with the diagnosis of FTLD and CBS and a large pedigree with autosomal dominant FTLD, for FUS and TDP-43 variants. In FUS we identified a spectrum of variants in patients and controls, while, for TDP-43, we report for the first time N267S in a case diagnosed with CBS. Our findings suggest that the pathogenicity of FUS variants needs to be revisited by examining the cosegregation and functional effect of those variants previously reported by other groups to be pathogenic and that variants need to be tested in larger cohorts of neurologically normal controls (preferably from the same population). Not least, it seems that genetic variability in TDP-43 encompasses different types of neurological disorders ranging from ALS to FTD and CBS.
Supplementary Material
Acknowledgments
Molecular genetics work was funded by the office of the Dean of the School of Medicine, Department of Internal Medicine, at Texas Tech Health Sciences Center and a grant from South Plains Foundation (PM). This work was also supported by NIH NINDS grant 5R00NS060766 (EDH), and the NINDS Intramural Research Program (JG). The authors thank Cynthia Crews, Anne Leopold, and Karen DeTucci for the study coordination, Dr Susan Bergeson for sharing her control DNA samples with PM for genetic screening, Poorna Dharmasri for proofreading of the manuscript, and Stephanie Cosentino for comments.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.neurobiolaging. 2011.08.004.
Footnotes
Disclosure statement
The authors disclose no conflicts of interest.
All human study protocols were approved by the ethics committee of local institutions. An informed consent was obtained from the persons with the power of attorney for the patient.
References
- Belzil VV, Valdmanis PN, Dion PA, Daoud H, Kabashi E, Noreau A, Gauthier J, S2D team. Hince P, Desjarlais A, Bouchard JP, Lacomblez L, Salachas F, Pradat PF, Camu W, Meininger V, Dupré N, Rouleau GA. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73:1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A. French Clinical Genetic Research Network on Frontotemporal Lobar Degeneration/Frontotemporal Lobar Degeneration with Motoneuron Disease 2009. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 65:470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- Boeve BF. Corticobasal degeneration: the syndrome and the disease. In: Litvan I, editor. Atypical Parkinsonian Disorders: Clinical and Research Aspects. Humana Press; Totowa, NJ: 2005. pp. 309–334. [Google Scholar]
- Boeve BF. The multiple phenotypes of corticobasal syndrome and corticobasal degeneration: implications for further study. J Mol Neurosci. 2011 Aug 19; doi: 10.1007/s12031-011-9624-1. [DOI] [PubMed] [Google Scholar]
- Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, Papetti A, Stuani C, Di Luca M, Gennarelli M, Padovani A. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat. 2009;30:E974–E983. doi: 10.1002/humu.21100. [DOI] [PubMed] [Google Scholar]
- Corrado L, Ratti A, Gellera C, Buratti E, Castellotti B, Carlomagno Y, Ticozzi N, Mazzini L, Testa L, Taroni F, Baralle FE, Silani V, D’Alfonso S. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat. 2009;30:688–694. doi: 10.1002/humu.20950. [DOI] [PubMed] [Google Scholar]
- Daoud H, Valdmanis PN, Kabashi E, Dion P, Dupré N, Camu W, Meininger V, Rouleau GA. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2009;46:112–114. doi: 10.1136/jmg.2008.062463. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Hardy J, Momeni P. Frontotemporal dementia: from Mendelian genetics towards genome wide association studies. J Mol Neurosci. 2011 Sep 6; doi: 10.1007/s12031-011-9635-y. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Kapogiannis D, Huey ED, Momeni P. FTD and ALS: a tale of two diseases. Curr Alzheimer Res. 2011b;8:273–294. doi: 10.2174/156720511795563700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Sleegers K, Engelborghs S, Robberecht W, Martin JJ, Vandenberghe R, Sciot R, Dermaut B, Goossens D, van der Zee J, De Pooter T, Del-Favero J, Santens P, De Jonghe P, De Deyn PP, Van Broeckhoven C, Cruts M. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging. 2009;30:1329–1331. doi: 10.1016/j.neurobiolaging.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kaplan H, Goodglass H, Weintraub S. Boston Naming Test. Lea & Febiger; Philadelphia: 1983. [Google Scholar]
- Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ, Budka H, Ghetti B, Spina S. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843–1847. doi: 10.1002/mds.22697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Bosco J, LeClerc AD, Tamrazian E, Van den Berg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;27:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Mattis S. Mental Status examination for organic mental syndrome in the elderly patient. In: Bellack L, Karusu TB, editors. Geriatric Psychiatry. Grune & Stratton; New York: 1976. pp. 77–121. [Google Scholar]
- Momeni P, Rogaeva E, Van Deerlin V, Yuan W, Grafman J, Tierney M, Huey E, Bell J, Morris CM, Kalaria RN, van Rensburg SJ, Niehaus D, Potocnik F, Kawarai T, Salehi-Rad S, Sato C, St George-Hyslop P, Hardy J. Genetic variability in CHMP2B and frontotemporal dementia. Neurodegener Dis. 2006;3:129–133. doi: 10.1159/000094771. [DOI] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Rollinson S, Snowden JS, Neary D, Morrison KE, Mann DM, Pickering-Brown SM. TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci Lett. 2007;419:1–4. doi: 10.1016/j.neulet.2007.03.044. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L, Rademakers R. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genetics. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher A, Friedrich P, Diehl-Schmid J, Ibach B, Perneczky R, Eisele T, Vukovich R, Foerstl H, Riemenschneider M. No association of TDP-43 with sporadic frontotemporal dementia. Neurobiol Aging. 2009;30:157–159. doi: 10.1016/j.neurobiolaging.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy DM, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijssens M, Peeters K, De Deyn PP, Cruts M, Van Broeckhoven C. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–371. doi: 10.1212/WNL.0b013e3181ccc732. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Memory Scale Revised. Harcourt Brace Jovanovich; San Antonio: 1987. [Google Scholar]
- Wechsler D. WAIS-III. Administration and Scoring Manual. Psychological Corporation; San Antonio: 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.