

Abstract
Specific point mutations in isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) occur in a variety of cancers, including acute myeloid leukemia (AML), low-grade gliomas, and chondrosarcomas. These mutations inactivate wild-type enzymatic activity and convey neomorphic function to produce D-2-hydroxyglutarate (D-2HG), which accumulates at millimolar levels within tumors. D-2HG can impact α-ketoglutarate-dependent dioxygenase activity and subsequently affect various cellular functions in these cancers. Inhibitors of the neomorphic activity of mutant IDH1 and IDH2 are currently in Phase I/II clinical trials for both solid and blood tumors. As IDH1 and IDH2 represent key enzymes within the tricarboxylic acid (TCA) cycle, mutations have significant impact on intermediary metabolism. The loss of some wild-type metabolic activity is an important, potentially deleterious and therapeutically exploitable consequence of oncogenic IDH mutations and requires continued investigation in the future. Here we review how IDH1 and IDH2 mutations influence cellular metabolism, epigenetics, and other biochemical functions, discussing these changes in the context of current efforts to therapeutically target cancers bearing these mutations.
Keywords: Metabolism, Mutant IDH1 and IDH2, Reductive carboxylation, Glucose oxidation, Mutant IDH inhibitors, NADPH
1. Introduction
Mutations in oncogenes and tumor suppressors facilitate the rapid growth of cancer cells and their survival in response to environmental stress. To maintain this phenotype tumor cells initiate a metabolic program that supplies the energy, reducing equivalents, and biosynthetic precursors necessary to divide (Tennant et al., 2010). In the early 20th century Otto Warburg observed that cancer cells (and normal proliferating cells) selectively metabolized glucose to lactate even under aerobic conditions (Warburg, 1956). This phenomenon, also known as the “Warburg Effect”, is common to many (but not all) tumors. Since Warburg’s discovery, biochemists have painstakingly annotated the network of biochemical reactions comprising cellular metabolism. Though not yet complete, this information provides a biochemical roadmap to study metabolic dysfunction in the context of diseases using a range of datasets (Bordbar et al., 2014; Yizhak et al., 2010). Most oncogenes and tumor suppressors directly impact cellular metabolism, and several hallmark cancer mutations have been observed to occur in metabolic enzymes. Homozygous loss-of-function mutations in fumarate hydratase (FH) or one of the five subunits comprising the succinate dehydrogenase (SDH) complex can lead to the development of specific cancers, representing the first time that metabolic enzymes were classified as bonafide tumor suppressors (King et al., 2006). More recently, mutations in isocitrate dehydrogenase 1 and 2 (IDH1, IDH2) have been discovered in various cancers. These exclusively heterozygous mutations do not follow a traditional loss-of-function mechanism, and the downstream effects of these mutations on tumor initiation, metabolism, and growth are currently being investigated. Here we review how mutations in IDH1 and IDH2 impact intermediary metabolism and other cell functions. Finally, the metabolic and epigenetic consequences of mutant IDH1 and IDH2 are discussed in the context of current efforts to therapeutically target cancers harboring these mutations.
2. Mutation of isocitrate dehydrogenase 1 and 2
Mutation of IDH1 and IDH2 were initially identified through exome sequencing of colon tumor and glioblastoma multiforme (GBM) (Parsons et al., 2008; Sjoblom et al., 2006). Since these discoveries, IDH mutations have been observed in several other tumor types, including acute myeloid leukemia (AML), chondrosarcoma, and intrahepatic cholangiocarcinoma (Amary et al., 2011; Borger et al., 2012; Mardis et al., 2009; Parsons et al., 2008; Yan et al., 2009). These mutations are somatically acquired and occur on distinct arginine residues of IDH1 (R132) and IDH2 (R172 or R140). Interestingly, IDH1 mutations occur at much higher incidences than IDH2 mutations in low grade gliomas, cholangiocarcinoma, and chondrosarcoma; however, IDH1 and IDH2 mutations occur at similar rates in AML (Molenaar et al., 2014; Ward et al., 2010). Due to the frequency of observation in low grade gliomas, IDH mutations are thought to play a significant role in early tumorigenesis and precede other oncogenic mutations (Balss et al., 2008; Juratli et al., 2013; Watanabe et al., 2009).
In contrast to SDH and FH mutants, which exhibit traditional homozygous loss-of-function mutations, IDH mutants retain one wild-type allele and rarely exhibit loss of heterozygosity (Jin et al., 2013; Mullen et al., 2012a). Furthermore, the occurrence of mutations on distinct IDH1 and IDH2 residues within the active site provided evidence that these changes elicit a gain-of-function phenotype in each enzyme. Subsequently, an analysis of the x-ray structure of mutant IDH1 in conjunction with metabolomics profiling demonstrated that (D)-2-hydroxyglutarate (D- or R-2HG) was produced by mutant IDH1 and accumulated at high levels in mutant tumors, confirming a gain-of-function mechanism (Dang et al., 2009). Similar production of D-2HG was demonstrated in cells and tumors harboring IDH2 mutations (Ward et al., 2010).
Wild-type IDH1 and IDH2 normally catalyze the reversible, NADP+-dependent oxidative decarboxylation of isocitrate to alpha-ketoglutarate in either the cytosol (IDH1) or mitochondria (IDH2). However, the mutant IDH enzyme loses oxidative activity and instead reduces alpha-ketoglutarate (aKG, also known as 2-oxoglutarate) to D-2HG, consuming one molecule of NADPH in the process (Figure 1). Under normal conditions human cells produce low levels of both D-2HG and L-2HG (or S-2HG) due to enzyme promiscuity, but 2HG (referring to both D-2HG and L-2HG) fails to accumulate due to the activity enantiomer-specific FAD-dependent 2-hydroxyglutarate dehydrogenases (L2HGDH and D2HGDH) that convert 2HG to aKG (Van Schaftingen et al., 2013). Deficiency in L2HGDH or D2HGDH due to homozygous loss-of-function mutation causes patients to develop 2HG aciduria characterized by an accumulation of either enantiomer in body fluids (Rzem et al., 2007; Struys, 2006). About 50% of patients with D-2HG aciduria have autosomal recessive mutations in D2HGDH; however, the majority of patients with normal D-2HGDH enzyme but high D-2HG harbored mutations in IDH2 (either R140Q or R140G) (Kranendijk et al., 2010). Patients with D-2HG aciduria either show no symptoms or exhibit developmental delay, epilepsy, cardiomyopathy, and other clinical symptoms (Kranendijk et al., 2010). In contrast, patients with L-2HG aciduria have an increased risk of developing certain brain cancers, suggesting that 2HG may act as a driver of tumorigenesis (DeBerardinis et al., 2012; Moroni et al., 2004). Most patients who developed metastatic brain tumors exhibited high levels of L-2HG, not D-2HG, and tumors that develop are of a different type than those commonly associated with IDH mutation (Cairns et al., 2013). Thus, D-2HG accumulation from mutant IDH may not be sufficient to drive malignancy and may require additional oncogenic mutations. Indeed, IDH mutations observed in low-grade gliomas frequently precede 1p/19q co-deletion and/or TP53 mutation which give rise to either oligoastrocytomas/oligodendrogliomas or low grade astrocytomas, respectively (Cairns et al., 2013; Ichimura, 2012; Labussiere et al., 2010; Lai et al., 2011). These tumors follow distinct transformation programs with 1p/19q co-deleted tumors commonly activating PI3K/Akt or Ras and p53 mutant tumors amplifying receptor tyrosine kinases (i.e., MET and PDGFR) (Wakimoto et al., 2014). Further transformation of IDH mutant low-grade gliomas into secondary glioblastomas requires EGFR amplification, PTEN loss, and/or additional genetic alterations (Lai et al., 2011).
Figure 1.
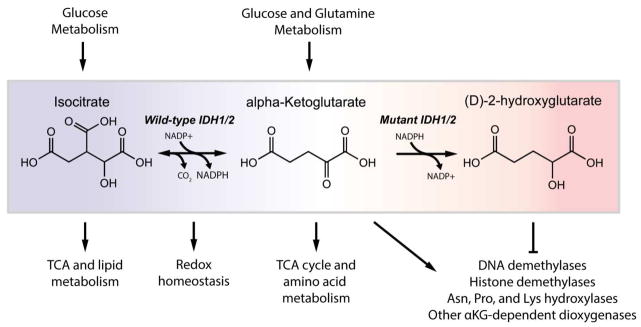
Multiple cellular pathways are affected by mutations in IDH1 and IDH2. Metabolites involved in these reactions are critical for glucose, glutamine, NADPH, amino acid, and lipid metabolism as well as epigenetic regulation.
Sequencing of IDH1 and IDH2 in AML patients indicated that these mutations occurred in a subset of tumors that were distinct from those harboring loss-of-function TET2 mutations, suggesting that D-2HG accumulation disrupts the function of TET2 or another aKG-dependent dioxygenase (Figueroa et al., 2010). Several studies have subsequently indicated that both L-2HG and D-2HG can act as competitive inhibitors of aKG-dependent dioxygenases, including the EglN family of prolyl hydroxylases (PHDs), the TET family of DNA demethylases, and the JmjC family of histone demethylases (Chowdhury et al., 2011; Koivunen et al., 2012; Lu et al., 2012; Xu et al., 2011). As such, D-2HG acts in a manner similar to the succinate and fumarate that accumulate in the context of SDH and FH mutant tumors, respectively (Selak et al., 2005; Xiao et al., 2012). Indeed, D-2HG accumulation resulting from mutant IDH expression has been observed to promote DNA and/or histone hypermethylation phenotypes (Figueroa et al., 2010; Lu et al., 2013; Turcan et al., 2012). At least in the context of SDH mutant cells, this inhibitory effect on dioxygenase activity can be ameliorated by addition of cell-permeable aKG analogs (MacKenzie et al., 2007). In addition, histone hypermethylation associated with IDH1 mutant expression in U87 glioma cells was reversed by octyl-aKG addition (Xu et al., 2011). In contrast to the inhibitory mechanisms noted above, D-2HG has been observed to activate EglN in many cell and in vivo models, leading to hypoxia inducible factor-1a (HIF1a) degradation and the promotion of tumor development (Koivunen et al., 2012; Losman et al., 2013b). On the other hand, other studies have observed increases in HIF1a levels in IDH1 mutant U87 cells (Xu et al., 2011; Zhao et al., 2009) or in brain-specific Nestin-IDH1R132H/wT transgenic mouse embryos (Sasaki et al., 2012a). In contrast, an analysis of IDH1-R132H and HIF1a expression in serial sections of IDH1-R132H positive gliomas suggested that IDH1-R132H expression was not sufficient for HIF1a stabilization (Williams et al., 2011). Overall, the role of IDH mutants and 2HG on HIF1a stabilization is complex and can be influenced by cell type, tissue, and the local microenvironment. The epigenetic dysregulation caused by aKG antagonism has been proposed to be one mechanism through which D-2HG contributes to tumorigenesis in mutant IDH tumors (Figure 1). However, the specific aKG-dependent dioxygenases that contribute to tumor development are likely to be context-dependent (e.g. tissue specific). As this family of enzymes catalyzes a wide variety of reactions and includes protein- and DNA-modifying enzymes as well as metabolic enzymes (reviewed by Losman & Kaelin, 2013), additional insights are needed to determine the mechanistic drivers of tumorigenesis downstream of mutant IDH (Losman et al., 2013a).
3. IDH mechanism and regulation
The crystal structures of human IDH1 and pig IDH2, which shares >97% identity with human IDH2, have yielded insights into the enzymatic and regulatory mechanisms of these NADP+-dependent enzymes (Ceccarelli et al., 2002; Xu et al., 2004). Structural studies of IDH1 suggest that its IDH1 follows a self-regulation feedback mechanism whereby isocitrate binds directly to Arg132, inducing a conformational change that allows the Asp279 residue to interact with Ca2+ cofactor and participate in catalysis (Xu et al., 2004). Kinetic studies suggest that isocitrate, and to a greater extent NADP+, regulate the activity and directionality of IDH1 (Rendina et al., 2013). The point mutations in IDH1 and IDH2 have significant effects on enzyme catalytic function and mechanism (Dang et al., 2009; Rendina et al., 2013). Arg132 directly interacts with isocitrate, and amino acid substitutions from any of the mutations observed in gliomas prevented isocitrate from binding (Dang et al., 2009; Zhao et al., 2009). Thus, IDH1 mutants become insensitive to physiological isocitrate levels and exhibit a >80% decreased capacity to carry out the oxidative reaction (Zhao et al., 2009). Consequently, the NADPH production by oxidative IDH activity is diminished, resulting in a ~38% reduction in the NADPH generation capacity in IDH1-mutant versus wild-type glioblastoma tumor tissue (Bleeker et al., 2010).
IDH1 mutants exhibit a sequential kinetic mechanism whereby NADPH first binds, reductively trapping aKG into D-2HG before allowing it to undergo carboxylation to form ICT (Rendina et al., 2013). IDH1-R132 variants (H, C, G, S, L) exhibit significantly different kinetic parameters for aKG and, consequently, produce different levels of D-2HG in cells expressing IDH1-R132 (Pusch et al., 2014). These mechanistic insights offer an explanation as to why D-2HG is preferentially produced by mutant IDH1 enzymes. In addition to the IDH1-R132 and IDH2-R172 and R140Q mutants, other IDH mutation sites have been predicted and/or demonstrated to exhibit neomorphic activity, including IDH1-R100, IDH1-Y179, and IDH1-G97 (Rendina et al., 2013; Ward et al., 2012). IDH1-Y179 and IDH1-G97 mutants exhibited lower Km values for isocitrate (i.e., improved binding); suggesting that neomorphic function is not reliant on an impaired utilization of isocitrate (Rendina et al., 2013). Ultimately, the changes in wild-type and neomorphic function of IDH1 and IDH2 described above influence cell signaling, epigenetics, and enzyme activity to directly and indirectly drive metabolic reprogramming within tumors (Figure 2).
Figure 2.

Biochemical pathways involved in intermediary metabolism. Glycolysis and glucose entry into the TCA cycle is regulated by the activity of lactate dehydrogenase (LDHA and LDHB), pyruvate dehydrogenase (PDH), and pyruvate carboxylation (PC). IDH1 and IDH2 are cytosolic and mitochondrial enzymes, respectively, that are critical for the metabolism of glucose- and glutamine- derived carbons. NADPH produced by either IDH1 or IDH2 is critical for maintaining the redox state in subcellular compartments. AcCoA: acetyl-coenzyme A, aKG: alpha-ketoglutarate, Cit: citrate, D-2HG: D-2-hydroxyglutarate, Fum: fumarate, ICT: isocitrate, Lac: lactate, Mal: malate, Oac: oxaloacetate, Pyr: pyruvate, Suc: succinate.
4. Glucose metabolism
Glycolytic flux is commonly upregulated in tumors downstream of various signaling pathways. For example, phosphoinositide 3-kinase (PI3K) is activated in many tumors and plays a significant role in maintaining the glycolytic phenotype of cancers through protein kinase B (PKB/Akt) signaling (Engelman, 2009). This oncogenic signal stimulates glycolysis, in part, by promoting the expression of glucose and other nutrient transporters and stimulating the activity of glycolytic enzymes including hexokinase and PFKFB3 (Cairns et al., 2011; DeBerardinis et al., 2008; Elstrom et al., 2004; Vander Heiden et al., 2009). While hyperactivation of PI3K/Akt signaling contributes to the aggressiveness of gliomas (Bleau et al., 2009; Koul, 2008), U87 glioma cells expressing IDH1-R132H exhibited decreased Akt levels at both the mRNA and protein level (Bralten et al., 2011). Furthermore, expression of mutant IDH1 in LN-319 glioblastoma cells caused a decrease in Akt phosphorylation, suggesting that mutant tumors may exhibit less of a glycolytic metabolic phenotype compared to IDH1 wild-type tumors (Birner et al., 2014). Importantly, the majority of tumors harboring both IDH mutations and 1p/19q co-deletion exhibit activation of PI3K/Akt; thus, the role of Akt on glucose metabolism in mutant IDH tumors may also rely on external factors (i.e. additional oncogenic mutations, tumor microenvironment).
Lactate dehydrogenase A (LDHA) is commonly upregulated in cancer cells downstream of HIF1a and Myc signaling (Cairns et al., 2011; Dang et al., 2008; Kim et al., 2007); this enzyme helps maintain glycolysis via NAD+ regeneration (Metallo et al., 2013). One recent study observed that IDH mutant gliomas and tumor-derived brain tumor stem cells silence LDHA expression through promoter hypermethylation (Chesnelong et al., 2014). In addition, one study identified that AML patients with IDH1 or IDH2 mutant tumors exhibited lower LDH activity compared to wild-type IDH1 or IDH2 tumors, suggesting a common phenotype between IDH mutants mediated by aKG/2HG (Chou et al., 2011). An analysis of gene expression in IDH1 mutant and IDH1 wild-type glioma samples demonstrated that factor inhibiting HIF-1 (FIH-1/HIFAN) was upregulated in IDH1 mutant tumors (Mustafa et al., 2014). FIH-1 inhibits the activity of HIF1a by preventing its transactivation in an aKG-dependent manner (Mahon et al., 2001), and HIF1a levels are known to be decreased in IDH1 mutant gliomas, enhancing tumorigenesis (Koivunen et al., 2012). This phenotype is thought to result from increased EglN activity fueled by 2HG-aKG isomerization (Tarhonskaya et al., 2014). Putatively as a consequence of HIF1a suppression, IDH1 mutant gliomas expressed high levels of LDHB relative to LDHA as compared to IDH1 wild-type gliomas or normal brain tissue (Mustafa et al., 2014). Acting as the final step in converting glucose to lactate, LDHA silencing may act to mitigate aerobic glycolysis. Although both isoforms of LDH (LDHA and LDHB) can metabolize the conversion of pyruvate to lactate, the LDHB isoform is more sensitive to substrate inhibition by pyruvate and is more capable of converting lactate to pyruvate (Dang, 2013).
Pyruvate dehydrogenase (PDH) is a major point of entry for glucose-derived pyruvate oxidation in the TCA cycle. PDH activity is regulated by its phosphorylation state and HIF1a stimulates expression of PDK1, leading to inactivating phosphorylation of PDH (Kim et al., 2006; Papandreou et al., 2006; Rardin et al., 2009; Semenza et al., 1994). Thus, alteration of HIF1a expression in mutant IDH cells and tumors may influence PDH activity and lead to changes in flux of glucose-derived pyruvate into the mitochondria. In addition to mitochondrial acetyl-CoA generated by PDH, cells need oxaloacetate (OAC) to maintain TCA cycle flux. Cancer cells can obtain OAC through various mechanisms, including glucose anaplerosis via pyruvate carboxylase or glutaminolysis, and pyruvate carboxylase is required for cells growing in glutamine-deprived conditions (Cheng et al., 2011). Furthermore, in vivo tracing studies in an orthotopic model of human glioblastoma using 13C-labeled glucose have indicated that pyruvate carboxylase and PDH are highly active in GBM (Marin-Valencia et al., 2012). One recent study demonstrated that IDH mutant overexpression in astrocytes results in an increased fractional flux through pyruvate carboxylase and an increase in PC expression, suggesting this pathway is critical for IDH mutant cells to maintain TCA activity (Izquierdo-Garcia et al., 2014). Consistent with this observation, we observed increased pyruvate cycling through malic enzymes and PC in HCT116 cells harboring heterozygous IDH1 mutations (Grassian et al., 2014).
5. Glutaminolysis, reductive carboxylation, and TCA metabolism
Glutamine is another major contributor to TCA metabolism in cancer cells and enters this pathway at aKG. As such, glutamine-glutamate-aKG metabolism represents a critical node in IDH mutant tumors. Glutamine is converted to glutamate during the biosynthesis of nucleotides, hexosamines, and asparagine; alternatively this reaction may be catalyzed in mitochondria via glutaminase (GLS). Transaminases or glutamate dehydrogenase (GLUD) can convert glutamate to the TCA intermediate and IDH substrate/product aKG. These pathways are highly active in most cancer cells as a result of oncogenic mutations or limited glucose oxidation (Gaglio et al., 2011; Son et al., 2013). Hypoxic microenvironments common to solid tumors promote glutamine flux into TCA metabolism such that it becomes the predominant carbon source for the glutaminolysis (Fan et al., 2013; Grassian et al., 2014; Le et al., 2012) and reductive carboxylation pathways (Metallo et al., 2012; Mullen et al., 2012b; Scott et al., 2011; Wise et al., 2011).
Not surprisingly, much of the D-2HG produced by mutant IDH1 and IDH2 in cells is derived from glutamine (Grassian et al., 2014). Due to its clinical prevalence and the availability of cell models more studies have focused on the impact of mutant IDH1 on TCA metabolism compared to mutant IDH2. Beyond aKG generation, flux through both glutaminolytic and reductive carboxylation pathways are significantly impacted by IDH mutations. When oxygen is replete, evidence suggests that IDH flux predominantly occurs in the oxidative direction, with minimal (but some) exchange observable. Given the impact of mutant IDH1 on WT activity it is not surprising that such cells become more reliant on the glutaminolysis pathway. Recent studies have highlighted differences in this pathway when cells express or harbor IDH1 mutations. For example, a glioblastoma cell line and transformed astrocytes both exhibited increased sensitivity to pharmacological or siRNA-mediated inhibition of glutaminase (Seltzer et al., 2010). In addition, Chen et al. recently observed that gliomas harboring IDH1 mutations overexpressed glutamate dehydrogenase 1 and 2 (GLUD1 and GLUD2), and orthotopic growth of mutant glioma lines were sensitive to GLUD1 or GLUD2 knockdown (Chen et al., 2014). We observed a similar increase in the dependence of IDH1 mutant cells on glutaminolysis in our analysis of a panel of HCT116 cells, providing evidence that these changes arise due to a direct impact on metabolism rather than indirectly through cell lineage-specific mechanisms (Grassian et al., 2014). Notably, this dependence on oxidative glutamine metabolism was exacerbated by culture under hypoxia, such that mutant IDH1 cells exhibited decreased growth and increased respiration under hypoxia (Grassian et al., 2014).
IDH1 has been implicated in catalyzing the reductive carboxylation of aKG to isocitrate, a pathway that facilitates conversion of glutamine to biosynthetic intermediates under conditions of hypoxia or mitochondrial dysfunction (Metallo et al., 2012; Mullen et al., 2012b; Scott et al., 2011). As the IDH reactions in human cells involve the interplay of NADH, NADPH, aKG, and isocitrate in two important cellular compartments, the localization, interconnectivity (i.e., via NAD(P)H shuttling), regulation, and function of the reductive carboxylation pathway is still actively investigated. In vitro enzyme studies have demonstrated that mutant/wild-type heterodimers of both IDH1 and IDH2 are unable to catalyze the reductive carboxylation reaction (Leonardi et al., 2012). Given the demonstrated role of IDH1 in this reaction, mutant IDH1 cells exhibit a strong defect in the conversion of glutamine to isocitrate, citrate, and acetyl-CoA under various conditions. Indeed, the extent that heterozygous mutant IDH HCT116 cells and IDH1-R132C HT1080 fibrosarcoma cells activate this pathway under hypoxia was compromised when compared to cells with IDH2 mutations or wild-type IDH (Grassian et al., 2014). Changes in glutamine metabolism under hypoxia were also observed in an additional study that employed HCT116 IDH1-R132H cells (Reitman et al., 2014). Furthermore, IDH1 mutant cells exhibited increased sensitivity to inhibitors of respiration, conditions known to promote reductive carboxylation (Fendt et al., 2013; Gameiro et al., 2013; Mullen et al., 2012b). This sensitivity could be due to the cells’ inability to synthesize acetyl-CoA through reductive carboxylation or alternatively due to their increased dependence on respiration under hypoxia. TCA metabolism is coupled with cellular respiration. As noted above, we observed increased sensitivity to ETC/respiration inhibitors and changes in oxygen consumption rates in IDH1 mutant HCT116 cells under hypoxia (Grassian et al., 2014). More recently, Chan et al. and other studies have demonstrated that D-2HG produced by mutant IDH inhibits complex IV (also known as cytochrome c oxidase, COX) of the ETC (Chan et al., 2015; da Silva et al., 2002; Latini et al., 2005; Wajne et al., 2002). This mechanism induced mutant IDH leukemia cell lines (patient-derived and engineered) to become sensitive to Bcl-2 inhibitors, initially identified as a target in a shRNA screen (Chan et al., 2015).
In addition to fatty acid and cholesterol synthesis, acetyl-CoA is an important building block for phospholipids, amino acids, and protein acetylation (Kaelin et al., 2013). Interestingly, N-acetylated amino acids including N-acetyl-aspartyl-glutamate (NAAG) and N-acetyl-aspartate (NAA) were significantly decreased in human glioma cells expressing IDH1-R132H (Reitman et al., 2011). These results suggest that mutant IDH tumors may exhibit perturbed acetyl-CoA metabolism, potentially due to changes in pathway fluxes fueling acetyl-CoA pools. In addition to differences in acetyl-CoA metabolism, IDH mutant tumors exhibited a significantly altered phospholipid profile compared to wild-type IDH tumors. Specifically, pools of the phospholipid metabolites phosphoethanolamine and glycerophosphocholine were significantly perturbed in mutant IDH versus wild-type IDH tumors (Esmaeili et al., 2014). Several of the oncogenic signaling pathways altered in IDH mutant tumors also impact fatty acid synthesis and uptake. For example, ATP-citrate lyase (ACL) acts as the major supplier of cytosolic AcCoA for fatty acid synthesis and is a major Akt substrate, and Akt also induces other fatty acid synthesis enzymes (i.e. FAS, ACC) via mTORC1 activation of SREBP-1 (Berwick et al., 2002; Porstmann et al., 2008; Ru et al., 2013). In addition to de novo synthesis, fatty acids and cholesterol can be scavenged from extracellular sources, and PI3K/Akt signaling can upregulate expression of the LDL receptor—supplying cells with cholesterol—via SREBP-1 (Guo et al., 2011). Of note, IDH1 is a transcriptional target of SREBP-1a and to a lesser extent SREBP-2, purportedly to supply NADPH for reductive biosynthesis in the cytosol (Shechter et al., 2003).
In part due to a lack of isogenic or cell-based models, fewer studies have addressed the impact of heterozygous mutations in IDH2 on intermediary metabolism. Generally, cells expressing IDH2-R172 accumulate more or similar amounts of D-2HG than those with mutant IDH1, though IDH2-R140Q mutants produce the least (Ward et al., 2010). In vitro enzyme studies and ectopic expression of mutants has indicated that differences in gene expression and compartment localization/conditions may influence the differential D-2HG production by IDH1-R132 mutants versus IDH2-R172 mutants (Ward et al., 2013). In addition to effects on D-2HG accumulation, some evidence indicates that the intermediary metabolism of cells with mutant IDH1 versus IDH2 differs as well. As noted above, detectable and significant effects on TCA metabolism were observed in an isogenic HCT116 cell panel cultured under normoxia and hypoxia. In contrast, the profile of glucose and glutamine-driven TCA metabolism in cells with either IDH2-R172 or IDH2-R140Q mutations was similar to that of parental cells cultured normally or in the presence of exogenous D-2HG (Grassian et al., 2014). No in vitro growth defect was observed under hypoxia, and the cells readily used the reductive carboxylation pathway for de novo lipogenesis. Similar trends were observed when comparing HT1080 (IDH1-R132C) fibrosacroma and SW1353 (IDH2-R172S) chondrosarcoma cells.
In the context of analyzing flux changes in heterozygous IDH mutant tumors versus those with WT IDH1 and IDH2 it is important to consider that a heterozygous mixture of homo- and heterodimers will exist within cells. Notably, the binding affinity of IDH1-R132 and IDH1-WT monomers was not significantly different, suggesting that a diversity of homo- and hetero- IDH1 dimers exists in IDH1 mutant tumors (Jin et al., 2011). In contrast, IDH2-R172 weakly binds IDH2-WT, indicating that there is a greater enrichment of WT-WT homodimers in mutant IDH2 tumors (Jin et al., 2011). Furthermore, differences in substrate availability for the IDH reaction are likely significant when comparing metabolism in the cytosol/peroxisome (IDH1) and mitochondrial matrix (IDH2). The ability to resolve such differences remains a challenge, as it is unclear to what extent reductive carboxylation is catalyzed in the mitochondria versus cytosol (Metallo et al., 2013). While defects in this pathway arise in mutant IDH1 cells but not mutant IDH2 cells, the expression of several mitochondrial enzymes (e.g. transhydrogenase, aKG-dehydrogenase complex) influences reductive carboxylation flux (Gameiro et al., 2013; Mullen et al., 2012b). Despite the observed in vitro differences in metabolism and growth, HCT116 cells containing either IDH1 or IDH2 mutations panel grew significantly slower as xenografts when compared to parental cells (Grassian et al., 2014). These findings highlight the importance of microenvironment on metabolism and the impact of IDH mutations as well as the need for better cell/tumor models.
6. Other metabolic pathways
Beyond glucose, glutamine, and acetyl-CoA metabolism, aKG and 2HG can influence a number of other metabolic pathways. As noted above, D-2HG may inhibit (or promote the activity of) other members of the aKG-dependent dioxygenase family. These enzymes catalyze diverse functions that include various metabolic reactions beyond demethylation (Losman et al., 2013a; Rose et al., 2011). For example, the activity of several proline and lysine hydroxylases are perturbed in the context of IDH mutations, leading to compromised collagen maturation and impacts on extracellular matrix (ECM) processing. Notably, a significant impact on ECM was observed in an IDH mutant knock-in model (Sasaki et al., 2012b). The dioxygenase family also includes enzymes involved in fatty acid metabolism, RNA modifications, and carnitine biosynthesis (Losman et al., 2013a; Rose et al., 2011). Furthermore, aKG is a substrate for a large number of enzymes outside of dioxygenases, including transaminases. Notably, BCAT1 expression is high in glioblastoma and suppressed by ectopic mutant IDH1 overexpression, suggesting that aKG or D-2HG levels influence branched chain amino acid (BCAA) metabolism (Tonjes et al., 2013). The activities of aspartate aminotransaminase (AST) and glutamate dehydrogenase (GDH), enzymes that utilize aKG as a substrate, were decreased in IDH1 mutant U87 cells due to changes in expression (AST) or posttranslational modifications (GDH) (Chaumeil et al., 2014). More focused, functional characterization of these pathways may highlight additional metabolic perturbations in IDH mutant tumors.
7. NADPH metabolism
In addition to the aforementioned TCA intermediates, the reactions catalyzed by IDH1 and IDH2 require NADPH as a cofactor (Figure 2). The pyridine nucleotide cofactor NADP(H) is critical for important cellular processes supporting redox homeostasis and biosynthesis of lipids and nucleotides (Pollak et al., 2007). NADP(H) exists as either the oxidized (NADP+) or reduced (NADPH) form, and the ratio of this redox couple heavily influences cellular physiology. The regeneration rate of reduced NADPH is extraordinarily high in proliferating cancer cells such that the pool turns over in approximately 20 minutes (Fan et al., 2014; Lewis et al., 2014). Classically, NADPH was thought to be regenerated primarily by the oxidative pentose phosphate pathway (PPP); however, recent evidence suggests that several other enzymes are also major contributors (Fan et al., 2014; Lewis et al., 2014; Pollak et al., 2007). These enzymes include malic enzymes (ME), isocitrate dehydrogenases (IDH), aldehyde dehydrogenases (ALDH), and methylene tetrahydrofolate dehydrogenases (MTHFD) (Lunt et al., 2011; Pollak et al., 2007; Tibbetts et al., 2010). Importantly, many of these enzymes have isoforms that exist in specific organelles (e.g., ME1 is cytosolic while ME2 and ME3 are mitochondrial), and since NADP(H) cannot transport directly across subcellular organelle membranes the maintenance of redox homeostasis in each organelle is distinctly regulated.
Mutation in either IDH1 or IDH2 deactivates the NADPH-production capacity of these enzymes; thus, mutant IDH cells may need to reroute flux through compensatory NADP+-dependent enzymes or suffer a decrease in available NADPH. In order to prevent oxidative damage from reactive oxygen species (ROS) generated during proliferation cells must maintain pools of reduced glutathione (GSH), the most abundant cellular antioxidant (Balendiran et al., 2004). Reduced glutathione can either be synthesized de novo or regenerated from oxidized glutathione (GSSG) via NADPH and glutathione reductase. A reduction in NADPH availability could lead to an increase in oxidative stress by decreasing GSH pools. In fact, one study of clonally selected cells overexpressing wild-type or IDH1-R132H glioma cells indicated that NADPH levels were decreased relative to wild-type IDH1 cells (Shi et al., 2014). Consequently, ROS and GSH levels were increased and decreased, respectively, in cells expressing mutant IDH1 (Shi et al., 2014). Furthermore, mutant IDH1 cells exhibited increased sensitivity to temozolomide (TMZ) and cis-diamminedichloroplatinum (CDDP), which can induce oxidative stress in tumor cells (Shi et al., 2014; SongTao et al., 2012). A similar sensitivity was observed when cells ectopically expressing mutant IDH1 or IDH2 were exposed to radiation (Li et al., 2013). These data provide some indication that oncogenic IDH1 perturbs NADPH homeostasis; however, the extent that these findings correlate with survival and treatment responsiveness remains unclear (Dubbink et al., 2009; Houillier et al., 2010). Ultimately, additional molecular studies are required to elucidate whether this increased sensitivity to oxidative stress is due to an inability to compensate metabolically or because of orthogonal effects of the mutation on cell physiology/epigenetics.
An in vivo knock-in model of IDH1-R132H exhibited increased NADP+/NADPH ratio, decreased GSH, and decreased ascorbate in whole brains, consistent with a decrease in NADPH production and redox control capacity (Sasaki et al., 2012a). However, intracellular ROS levels in total brains of IDH1-R132H knock-in mice were significantly reduced relative to IDH1-WT knock-in brains (Sasaki et al., 2012a). Inhibition of IDH1-R132H may increase total ROS levels and, along with reduced NADPH and GSH levels, increase oxidative stress in these tumors and lead to cell death. High levels of ROS can damage lipids, proteins, and DNA and can lead to the activation of apoptosis and disruption of the cell cycle (Finkel et al., 2000). In addition, high levels of mitochondrial ROS may contribute significantly to mitochondrial dysfunction, as mtDNA is more readily damaged than nuclear DNA (Kim et al., 2015). Mutation of mtDNA has been observed to contribute to tumorigenicity in several cancer types (Sabharwal et al., 2014); to this end, mutant IDH2 cells in particular may exhibit compromised mitochondrial NADPH homeostasis, which may lead to increased mtDNA mutation and mitochondrial dysfunction.
8. Allelic inhibitors of mutant IDH1 and IDH2
Given the distinct, gain-of-function activity caused by IDH mutations, several efforts have identified selective pharmacological agents that target mutant IDH1 and IDH2 enzymes. One of the first compounds (AGI-5198) to be discovered specifically inhibited IDH1-R132H and IDH1-R132C mutant enzymes, reduced 2HG levels in glioma cells, and impaired growth of IDH1 mutant but not IDH1 WT glioma xenografts (Rohle et al., 2013). AGI-5198 suppression of 2HG levels did not completely ameliorate the DNA hypermethylation phenotype in mutant IDH1 glioma cells, suggesting that mutant IDH1-mediated epigenetic dysregulation is not easily reversed (Rohle et al., 2013). Since AGI-5198 was discovered, several other inhibitors have been identified that reduce D-2HG production in both in vitro and in vivo models (Popovici-Muller et al., 2012; Zheng et al., 2013). Shortly after the discovery of AGI-5198, medicinal chemistry optimization yielded the first inhibitor of IDH2-R140Q (AGI-6780) that reversed the hematopoietic differentiation induced by IDH2-R140Q in TF-1 erythroleukemia cells (Wang et al., 2013). Several additional inhibitors of the IDH2-R172K and IDH2-R140Q, the two highest frequency IDH2 mutations, have recently been discovered, including IDH2-C100 and AG-221, a derivative of AGI-6780, which both exhibit efficacy in cell and in vivo models (Patent WO 2013102431) (Yen et al., 2013). In aggressive IDH2 mutant primary AML xenografts models, AG-221 treatment reduced 2HG levels >90%, reversed histone and DNA hypermethylation, and conferred significant survival benefits to mice (Yen et al., 2013).
Clinical data from Phase I/II trials are emerging at a rapid rate, providing encouraging results for AML. As we continue to gain a better appreciation of the response of solid and blood tumors to these inhibitors, alternative approaches worth investigating are combinatorial treatments that target the metabolic deficiencies in IDH mutant tumors. While resistance to changes the epigenetic state of IDH mutant cells may emerge, these tumors are unlikely to regain the wild-type IDH1 or IDH2 activity that was originally lost to mutation. Therefore, pharmacological inhibition of the specific metabolic pathways on which IDH1 or IDH2 mutant cells are critically dependent may prove efficacious.
9. Conclusion
The discovery, functional characterization, and clinical development of therapies surrounding oncogenic IDH mutations highlight the great potential impact of advanced scientific technologies on medicine. In order to fully exploit the metabolic and physiological defects of IDH mutant tumors additional studies are required to identify and target such biochemical pathways in cellular and preclinical models. Improved biological models are still required, since patient-derived IDH mutant tumor cells grow slowly, ectopic expression of mutant enzymes is unstable and ineffective at producing high D-2HG levels, and isogenic, engineered cell lines lack appropriate biological context. Ultimately, molecular level analyses of how IDH mutations impact the metabolism, epigenetics, and oncogenic development of tumors will lead to additional insights into the pathogenesis of other transforming events (e.g. SDH and FH-deficient tumors) and inborn errors of metabolism (L-2HG and D-2HG aciduria).
Abbreviations
- ACC
acetyl-CoA carboxylase
- ACO
aconitase
- AML
acute myeloid leukemia
- aKG
alpha-ketoglutarate
- D-2HG
D-2-hydroxyglutarate
- FAS
fatty acid synthase
- Fum
fumarate
- FH
fumarate hydratase
- ICT
isocitrate
- IDH
isocitrate dehydrogenase
- L-2HG
L-2-hydroxyglutarate
- LDH
lactate dehydrogenase
- Mal
malate
- Oac
oxaloacetate
- Pyr
pyruvate
- PC
pyruvate carboxylase
- PDH
pyruvate dehydrogenase
- SDH
succinate dehydrogenase
Footnotes
Conflict of Interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224(3):334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct. 2004;22(6):343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116(6):597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277(37):33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- Birner P, Pusch S, Christov C, Mihaylova S, Toumangelova-Uzeir K, Natchev S, et al. Mutant IDH1 inhibits PI3K/Akt signaling in human glioma. Cancer. 2014;120(16):2440–2447. doi: 10.1002/cncr.28732. [DOI] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119(4):487–494. doi: 10.1007/s00401-010-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordbar A, Monk JM, King ZA, Palsson BO. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet. 2014;15(2):107–120. doi: 10.1038/nrg3643. [DOI] [PubMed] [Google Scholar]
- Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17(1):72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bralten LB, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011;69(3):455–463. doi: 10.1002/ana.22390. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3(7):730–741. doi: 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- Ceccarelli C, Grodsky NB, Ariyaratne N, Colman RF, Bahnson BJ. Crystal structure of porcine mitochondrial NADP+-dependent isocitrate dehydrogenase complexed with Mn2+ and isocitrate. Insights into the enzyme mechanism. J Biol Chem. 2002;277(45):43454–43462. doi: 10.1074/jbc.M207306200. [DOI] [PubMed] [Google Scholar]
- Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil MM, Larson PE, Woods SM, Cai L, Eriksson P, Robinson AE, et al. Hyperpolarized [1–13C] glutamate: a metabolic imaging biomarker of IDH1 mutational status in glioma. Cancer Res. 2014;74(16):4247–4257. doi: 10.1158/0008-5472.CAN-14-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci U S A. 2014;111(39):14217–14222. doi: 10.1073/pnas.1409653111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108(21):8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnelong C, Chaumeil MM, Blough MD, Al-Najjar M, Stechishin OD, Chan JA, et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014;16(5):686–695. doi: 10.1093/neuonc/not243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. 2011;25(2):246–253. doi: 10.1038/leu.2010.267. [DOI] [PubMed] [Google Scholar]
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva CG, Ribeiro CA, Leipnitz G, Dutra-Filho CS, Wyse AA, Wannmacher CM, et al. Inhibition of cytochrome c oxidase activity in rat cerebral cortex and human skeletal muscle by D-2-hydroxyglutaric acid in vitro. Biochim Biophys Acta. 2002;1586(1):81–91. doi: 10.1016/s0925-4439(01)00088-6. [DOI] [PubMed] [Google Scholar]
- Dang CV. Role of aerobic glycolysis in genetically engineered mouse models of cancer. BMC Biol. 2013;11:3. doi: 10.1186/1741-7007-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8(1):51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbink HJ, Taal W, van Marion R, Kros JM, van Heuvel I, Bromberg JE, et al. IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology. 2009;73(21):1792–1795. doi: 10.1212/WNL.0b013e3181c34ace. [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- Esmaeili M, Hamans BC, Navis AC, van Horssen R, Bathen TF, Gribbestad IS, et al. IDH1 R132H mutation generates a distinct phospholipid metabolite profile in glioma. Cancer Res. 2014;74(17):4898–4907. doi: 10.1158/0008-5472.CAN-14-0008. [DOI] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:712. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510(7504):298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73(14):4429–4438. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17(3):372–385. doi: 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74(12):3317–3331. doi: 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1(5):442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75(17):1560–1566. doi: 10.1212/WNL.0b013e3181f96282. [DOI] [PubMed] [Google Scholar]
- Ichimura K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 2012;29(3):131–139. doi: 10.1007/s10014-012-0090-4. [DOI] [PubMed] [Google Scholar]
- Izquierdo-Garcia JL, Cai LM, Chaumeil MM, Eriksson P, Robinson AE, Pieper RO, et al. Glioma cells with the IDH1 mutation modulate metabolic fractional flux through pyruvate carboxylase. PLoS One. 2014;9(9):e108289. doi: 10.1371/journal.pone.0108289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Reitman ZJ, Duncan CG, Spasojevic I, Gooden DM, Rasheed BA, et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013;73(2):496–501. doi: 10.1158/0008-5472.CAN-12-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Reitman ZJ, Spasojevic I, Batinic-Haberle I, Yang J, Schmidt-Kittler O, et al. 2-hydroxyglutarate production, but not dominant negative function, is conferred by glioma-derived NADP-dependent isocitrate dehydrogenase mutations. PLoS One. 2011;6(2):e16812. doi: 10.1371/journal.pone.0016812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juratli TA, Peitzsch M, Geiger K, Schackert G, Eisenhofer G, Krex D. Accumulation of 2-hydroxyglutarate is not a biomarker for malignant progression in IDH-mutated low-grade gliomas. Neuro Oncol. 2013;15(6):682–690. doi: 10.1093/neuonc/not006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153(1):56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Won SJ, Fabian C, Kang M, Szardenings M, Shin M. Mitochondrial DNA Aberrations and Pathophysiological Implications in Hematopoietic Diseases, Chronic Inflammatory Diseases, and Cancers. Ann Lab Med. 2015;35(1):1–14. doi: 10.3343/alm.2015.35.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27(21):7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25(34):4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul D. PTEN signaling pathways in glioblastoma. Cancer Biol Ther. 2008;7(9):1321–1325. doi: 10.4161/cbt.7.9.6954. [DOI] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330(6002):336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- Labussiere M, Idbaih A, Wang XW, Marie Y, Boisselier B, Falet C, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74(23):1886–1890. doi: 10.1212/WNL.0b013e3181e1cf3a. [DOI] [PubMed] [Google Scholar]
- Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. 2011;29(34):4482–4490. doi: 10.1200/JCO.2010.33.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini A, da Silva CG, Ferreira GC, Schuck PF, Scussiato K, Sarkis JJ, et al. Mitochondrial energy metabolism is markedly impaired by D-2-hydroxyglutaric acid in rat tissues. Mol Genet Metab. 2005;86(1–2):188–199. doi: 10.1016/j.ymgme.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem. 2012;287(18):14615–14620. doi: 10.1074/jbc.C112.353946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell. 2014;55(2):253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Chou AP, Chen W, Chen R, Deng Y, Phillips HS, et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 2013;15(1):57–68. doi: 10.1093/neuonc/nos261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013a;27(8):836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013b;339(6127):1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Venneti S, Akalin A, Fang F, Ward PS, Dematteo RG, et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27(18):1986–1998. doi: 10.1101/gad.226753.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, et al. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol. 2007;27(9):3282–3289. doi: 10.1128/MCB.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15(20):2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15(6):827–837. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481(7381):380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49(3):388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar RJ, Radivoyevitch T, Maciejewski JP, van Noorden CJ, Bleeker FE. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim Biophys Acta. 2014;1846(2):326–341. doi: 10.1016/j.bbcan.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Moroni I, Bugiani M, D’Incerti L, Maccagnano C, Rimoldi M, Bissola L, et al. L-2-hydroxyglutaric aciduria and brain malignant tumors: a predisposing condition? Neurology. 2004;62(10):1882–1884. doi: 10.1212/01.wnl.0000125335.21381.87. [DOI] [PubMed] [Google Scholar]
- Mullen AR, DeBerardinis RJ. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol Metab. 2012a;23(11):552–559. doi: 10.1016/j.tem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012b;481(7381):385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa DA, Swagemakers SM, Buise L, van der Spek PJ, Kros JM. Metabolic alterations due to IDH1 mutation in glioma: opening for therapeutic opportunities? Acta Neuropathol Commun. 2014;2:6. doi: 10.1186/2051-5960-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak N, Dolle C, Ziegler M. The power to reduce: pyridine nucleotides--small molecules with a multitude of functions. Biochem J. 2007;402(2):205–218. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F, et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med Chem Lett. 2012;3(10):850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8(3):224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch S, Schweizer L, Beck AC, Lehmler JM, Weissert S, Balss J, et al. D-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol Commun. 2014;2:19. doi: 10.1186/2051-5960-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem. 2009;389(2):157–164. doi: 10.1016/j.ab.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan LJ, Gooden DM, et al. Cancer-associated isocitrate dehydrogenase 1 (IDH1) R132H mutation and d-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J Biol Chem. 2014;289(34):23318–23328. doi: 10.1074/jbc.M114.575183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108(8):3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendina AR, Pietrak B, Smallwood A, Zhao H, Qi H, Quinn C, et al. Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry. 2013;52(26):4563–4577. doi: 10.1021/bi400514k. [DOI] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40(8):4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- Ru P, Williams TM, Chakravarti A, Guo D. Tumor metabolism of malignant gliomas. Cancers (Basel) 2013;5(4):1469–1484. doi: 10.3390/cancers5041469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis. 2007;30(5):681–689. doi: 10.1007/s10545-007-0487-0. [DOI] [PubMed] [Google Scholar]
- Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14(11):709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012a;26(18):2038–2049. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012b;488(7413):656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286(49):42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269(38):23757–23763. [PubMed] [Google Scholar]
- Shechter I, Dai P, Huo L, Guan G. IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: evidence that IDH1 may regulate lipogenesis in hepatic cells. J Lipid Res. 2003;44(11):2169–2180. doi: 10.1194/jlr.M300285-JLR200. [DOI] [PubMed] [Google Scholar]
- Shi J, Sun B, Shi W, Zuo H, Cui D, Ni L, et al. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol. 2014 doi: 10.1007/s13277-014-2644-z. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SongTao Q, Lei Y, Si G, YanQing D, HuiXia H, XueLin Z, et al. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012;103(2):269–273. doi: 10.1111/j.1349-7006.2011.02134.x. [DOI] [PubMed] [Google Scholar]
- Struys EA. D-2-Hydroxyglutaric aciduria: unravelling the biochemical pathway and the genetic defect. J Inherit Metab Dis. 2006;29(1):21–29. doi: 10.1007/s10545-006-0317-9. [DOI] [PubMed] [Google Scholar]
- Tarhonskaya H, Rydzik AM, Leung IK, Loik ND, Chan MC, Kawamura A, et al. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat Commun. 2014;5:3423. doi: 10.1038/ncomms4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr. 2010;30:57–81. doi: 10.1146/annurev.nutr.012809.104810. [DOI] [PubMed] [Google Scholar]
- Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013;19(7):901–908. doi: 10.1038/nm.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schaftingen E, Rzem R, Marbaix A, Collard F, Veiga-da-Cunha M, Linster CL. Metabolite proofreading, a neglected aspect of intermediary metabolism. J Inherit Metab Dis. 2013;36(3):427–434. doi: 10.1007/s10545-012-9571-1. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajne M, Vargas CR, Funayama C, Fernandez A, Elias ML, Goodman SI, et al. D-2-Hydroxyglutaric aciduria in a patient with a severe clinical phenotype and unusual MRI findings. J Inherit Metab Dis. 2002;25(1):28–34. doi: 10.1023/a:1015165212965. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014;20(11):2898–2909. doi: 10.1158/1078-0432.CCR-13-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–270. [PubMed] [Google Scholar]
- Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, et al. Identification of additional IDH mutations associated with oncometabolite R(−)-2-hydroxyglutarate production. Oncogene. 2012;31(19):2491–2498. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Lu C, Cross JR, Abdel-Wahab O, Levine RL, Schwartz GK, et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem. 2013;288(6):3804–3815. doi: 10.1074/jbc.M112.435495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174(4):1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SC, Karajannis MA, Chiriboga L, Golfinos JG, von Deimling A, Zagzag D. R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1alpha upregulation in adult glioma. Acta Neuropathol. 2011;121(2):279–281. doi: 10.1007/s00401-010-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108(49):19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, et al. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004;279(32):33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Wang F, Travins J, Chen Y, Hua Y, Straley K, et al. AG-221 Offers a Survival Advantage In a Primary Human IDH2 Mutant AML Xenograft Model. Blood. 2013;122(21) [Google Scholar]
- Yizhak K, Benyamini T, Liebermeister W, Ruppin E, Shlomi T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics. 2010;26(12):i255–260. doi: 10.1093/bioinformatics/btq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Yao Y, Liu Z, Deng L, Anglin JL, Jiang H, et al. Crystallographic Investigation and Selective Inhibition of Mutant Isocitrate Dehydrogenase. ACS Med Chem Lett. 2013;4(6):542–546. doi: 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]