

Abstract
The AMPK-related kinase NUAK2 has been implicated in melanoma growth and survival outcomes but its therapeutic utility has yet to be confirmed. In this study, we show how its genetic amplication in PTEN-deficient melanomas may rationalize the use of CDK2 inhibitors as a therapeutic strategy. Analysis of array-CGH data revealed that PTEN deficiency is coupled tightly with genomic amplification encompassing the NUAK2 locus, a finding strengthened by immunohistochemical evidence that phospho-Akt overexpression was correlated with NUAK2 expression in clinical specimens of acral melanoma. Functional studies in melanoma cells showed that inactivation of the PI3K pathway upregulated p21 expression and reduced the number of cells in S phase. NUAK2 silencing and inactivation of the PI3K pathway efficiently controlled CDK2 expression, whereas CDK2 inactivaiton specifically abrogated the growth of NUAK2-amplified and PTEN-deficient melanoma cells. Immunohistochemical analyses confirmed an association of CDK2 expression with NUAK2 amplification and p-Akt expression in melanomas. Lastly, pharmacological inhibition of CDK2 was sufficient to suppress the growth of NUAK2-amplified and PTEN-deficient melanoma cells in vitro and in vivo. Overall, our results identify show how CDK2 blockade may offer a promising therapy for genetically-defined melanomas where NUAK2 is amplified and PTEN is deleted.
Keywords: NUAK2, PTEN, PI3K pathway, CDK2, cell cycle, melanoma
Introduction
Recent advances in cancer genomics facilitate the elucidation of aberrant downstream pathways in tumor cells with genomic aberrations and pave the way to develop specific therapies for novel oncogenes and tumor-suppressor genes in many types of cancers (1–4). In melanomas, several genomic aberrations, such as mutations, amplifications and deletions in BRAF, NRAS, INK4A, MITF, PREX2, GNAQ and KIT, have been reported, and recent analyses using array-CGH data also suggested that NUAK2, which resides at chromosome 1q32, is an important gene that regulates cell cycle progression and cell migration in melanoma cells (2;5–14). The significance of NUAK2 in melanomagenesis is highlighted by the fact that high expression of NUAK2 has an impact on the survival of patients with acral melanomas in addition to the fact that NUAK2 participates in the regulation of cell proliferation of melanomas in general (12). On the other hand, synergistic effects of several genomic aberrations are also quite important to facilitate tumorigenesis of cancer cells such as that the PI3K pathway participates in melanomagenesis (15;16).
The elucidation of genomic aberrations including mutations has progressed using systematic approaches (17). However, detailed mechanisms controlling cell cycle progression by NUAK2 and additional genes remain to be elucidated. Analyses of cell cycle progression in NRAS-mutated and MITF-amplified melanomas showed that control of the cell cycle is differently regulated by CDKs in melanoma cells, where CDK4 is a key driver in NRAS-mutant melanomas, while CDK2 has a pivotal role in melanomas with high expression of MITF (18–20) Those results imply that elucidation of mechanisms regulating the cell cycle by different genomic aberrations should reveal the different impact of CDKs on the cell cycle.
In melanomas, BRAF mutations have been identified as activating mutations that facilitate melanomagenesis, and this discovery accelerated molecular targeted therapies against melanomas using drugs such as vemurafenib and dabrafenib (1;2). However BRAF mutations have diverse discrepancies among subtypes of melanomas (21). Some subtypes of melanomas, such as acral and mucosal melanomas, have low frequencies of BRAF mutations and are speculated to respond poorly to those therapies targeting BRAF mutations. Molecular targeted therapies aimed at genomic aberrations other than BRAF mutations should be developed for better management of patients with those subtypes of melanomas.
In this study, we explore additional genomic aberrations and downstream pathways of NUAK2, and demonstrate that NUAK2 and the PI3K pathway coordinately control CDK2. In addition, we showed that CDK2 is an efficient therapeutic target by abrogating the growth of cutaneous melanomas.
Material and Methods
Tumor specimens
We obtained 91 paraffin-embedded specimens of primary melanomas from 3 Institutions. This study was approved by the Tokyo Medical and Dental University Research Committee, the Osaka University Clinical Research Committee and the Saitama Cancer Center Research Ethics Committee. Fifty-six tumors were classified as acral melanomas and 35 as non-chronic sun-induced damage (CSD) melanomas, but none was a CSD melanoma according to the definition by Curtin and colleagues (22).
Cell lines
Normal human melanocyte and melanoma cell lines were cultured and maintained as previously described (23). C32, A375 and Malme-3M melanoma cells were purchased from the American Type Culture Collection (Manassas, VA). SKMel28 and SKMel23 melanoma cells were kindly provided by the Surgery Branch, NCI/NIH (Bethesda, MD). SM2-1 melanoma cells were kindly provided by Dr. H. Murata (Shinshu University, Matsumoto, Japan). The Mel2 melanoma cell line was established from a lymph node metastasis of a 68-year-old Japanese male acral melanoma patient in 1998 and the mel18 melanoma cell line was established from a lymph node metastasis of a 51-year-old Japanese male acral melanoma patient in 1998 in our laboratory, as described previously (24). C32, mel2, mel18 and SM2-1 melanoma cell lines were cultured in RPMI1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 IU/mL penicillin and 100 μg/mL streptomycin at 37ºC in a 5% CO2 incubator. All other melanoma cells were cultured in DMEM with 5% FBS. The original C32 , A375 and Malme-3M melanoma cells were STR DNA profiled in 2012.
Vectors, siRNA transfection and Lentiviral infection
SMARTpool siRNAs against CDK2 and PTEN were purchased from Thermo Fisher Scientific (Waltham, MA). Lentiviral vectors carrying shRNA targeting NUAK2 (AAB66-F-6: AAACCCAGGGCTGCCTTGGAAAAG and AAB66-F-7: AAACCCAGGGCTGCCTTGGAAAAG) and the empty vector were purchased from Open Biosystems (Rockford, IL) in the pLKO.1puro vector. For siRNA experiments, cells were seeded at 3.0 × 105 cells/well in 6-well plates and were transfected either with an siNT (non-targeting) or with an siRNA against CDK2 or PTEN (SMARTpool siRNAs, Thermo Fisher Scientific) at a concentration of 100 pmol/well using Lipofectamine RNAiMAX (Invitrogen, Grand Island, NY) according to the manufacturer’s protocol. All siRNA experiments were performed in triplicate. Infection of Lentivirus containing shRNA constructs with pLKO.1 against NUAK2 into cells were performed as previously described (12).
In Vitro Assays
For cell number analyses using siRNA, cells were seeded at 3.0 × 105 cells/well in 6-well plates in triplicate. Cell numbers were counted at Day0, Day2 and Day4 after transfection of siRNA. For cell number analyses treated with Roscovitine, cells were seeded at 2.0 × 105 cells/well in 6-well plates. Cell numbers were counted at Day3, Day5 and Day7 after treatment with Roscovitine.
For proliferation assays of C32 and SM-KT1 cells, cells were seeded at 1.0 × 105 cells/well in 24-well plates in quadruplicate. At 48 h, cell proliferation were measured using the MTS assay according to the manufacturer’s protocol (Takara Bio, Shiga, Japan).
For colony growth assays with Roscovitine, cells were seeded at 1.0 × 105 cells/well (C32, mel2 and mel18) or 5.0 × 104 cells/well (A375, SKMel28 and SKMel23) in 6-well plates in triplicate. After treatment with Roscovitine for 14 days, cells were fixed and stained with crystal violet; measurements were performed at an optical density of 610 nm.
Cell cycle profile analyses were performed as previously described (12). Cells were treated with Ly294002 at 20 μM for 24 h.
Animal model
All animal experiments were approved by the Animal Care and Use Committee of the Keio University. Three × 106 C32 melanoma cells, 3.0 × 106 SM2-1 melanoma cells and 3.0 × 106 mel18 melanoma cells were injected subcutaneously into nude mice (4 or 5 per group as noted). Seven days after injection of tumor cells, mice were orally treated with 2 mg/dose Roscovitine (every day for 10 days). Tumor sizes were then measured at day 10 of treatment.
Immunoblotting
Immunoblotting was performed as previously described (20). Antibodies used included a rabbit monoclonal anti-phospho(Ser473) Akt antibody (1:1000, Cell Signaling, Danvers, MA), a rabbit polyclonal anti-NUAK2 antibody (1:1000, Proteintech Group, Chicago, IL), a mouse monoclonal anti-actin antibody (1:1000; Abcam, Cambridge, MA), a rabbit polyclonal anti-CDK2 antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA), a rabbit monoclonal anti-CDK4 antibody (1:1000, Cell Signaling), a mouse monoclonal anti-CDK6 antibody (1:500, Abcam), a rabbit monoclonal anti-p21 antibody (1:1000, Cell Signaling), a rabbit monoclonal anti-p27 antibody (1:1000, Abcam), a rabbit monoclonal anti-Akt(pan) antibody (1:1000, Cell Signaling) and a rabbit monoclonal anti-PTEN antibody (1:1000, Cell Signaling).
Immunohistochemical analysis
Immunohistochemistry was performed as previously described (12). Antibodies were used at the following dilutions: anti-phospho(Ser473)Akt (1:25), anti-PTEN (1:100), anti-NUAK2 (1:100), anti-p27 (1:100) and anti-CDK2 (1:2000). For immunostaining of CDK2 and p27, staining was developed with a Vector VIP Substrate Kit (Vector Laboratories, Burlingame, CA) in pigmented melanomas or with a Vector DAB Substrate Kit (Vector Laboratories) using counter-staining in non-pigmented melanomas. Cells stained in the nucleus and/or both the nucleus and cytoplasm were counted as positive, and cells that stained only in the cytoplasm were counted as negative. Immunostaining of CDK2 was scored from 0 to +3 (0 = 0 to10%, +1 =11% to 25%, +2 = 26% to 50% or +3 = 51% to 100%) depending on the percentages of cells in a blind fashion by 3 observers. The basal expression group (negative staining group) includes specimens with a 0 score and the over-expression group (positive staining group) includes specimens with +1, +2 or +3 scores.
Immunohistochemistry using fluorescence was performed as previously described (12) using the anti-p21 antibody (1:100). Images were captured using a Leica DMR B/D MLD fluorescence microscope (Leica, Weltzar, Germany) and a Dage-MTI 3CCD 3-chip color video camera (Dage-MTI, Michigan City, IN).
Statistical analysis
For comparisons between two groups, P values were calculated using two-tailed Student’s t tests. Repeated two way ANOVA analysis was applied to check the drug effect of Roscovitine on C32 cells and mel18 cells. In all experiments, differences were considered statistically significant at P<0.05. Statistical analyses were performed using SAS 9.2. Fisher's exact test was used to test the relationship between NUAK2 expression and p-AKT(S473) expression in acral melanoma and Non-CSD patients. The differences of survival time among patients with different types of gene expression were tested by log rank test. Kaplan Meier curves were performed in the R Survival package.
Results
Downstream Pathways affected by NUAK2
To efficiently explore downstream targets that could suppress the growth of NUAK2-amplified melanoma cells, we set out to elucidate additional genomic changes and mechanisms that would facilitate the identification of targets in the downstream pathway of NUAK2. We first explored potential correlations between NUAK2 amplification and genetic aberrations of other melanoma-related genes: CDKN2A deletion, CDK4 gain, MDM2 gain, CCND1 gain and PTEN deletion using a public array database (Series GSE2631). Biostatistical analysis showed that only a deletion of PTEN correlated with the gain of ”RP11-243M13”, which is the nearest clone to NUAK2 (P = 0.0004), in acral melanomas. To confirm this, we used immunohistochemical analyses of clinical specimens to show that the expression of NUAK2 and p-Akt(S473) has a significant correlation (P < 0.001) in acral melanomas (Table 1, Supplementary Fig. S1 and S2, and Table S1). Kaplan Meier curves were used to show survival time differences among patients with different types of gene expression, and relapse free survival time of patients with expression of both NUAK2 and p-Akt(S473) were dramatically shorter than patients with either NUAK2 or p-Akt alone (P = 0.002) compared to overall survival time (P = 0.072) (Fig. 1A and Supplementary Fig. S3A).Survival analysis were also applied to data of Non-CSD melanoma patients but no significant differences were found among patients with different gene expression types (Fig.1B and Supplementary Fig. S3B). Those analyses led us to speculate that cooperation between both the NUAK2 and PI3K pathways is critical to tumorigenesis and that convergent points of those two pathways would be efficient targets to suppress the growth of NUAK2-amplified melanomas.
Table 1. Relationship of NUAK2 and p-Akt(S473) expression by immunohistochemical analyses using clinical specimens.
The contingency table displays variable values. Fisher’s exact test shows that the expression of NUAK2 and p-Akt(S473) has a significant correlation (P < 0.001) in acral melanomas.
| frequency | p-Akt(S473) expression | |||
|---|---|---|---|---|
| positive | negative | total | ||
| NUAK2 expression | positive | 29(51.97%) | 2 (3.57%) | 31 (55.36%) |
| negative | 12(21.43%) | 13(23.21%) | 25 (44.64%) | |
| total | 41 (73.21%) | 15(26.79%) | 56(100.00%) | |
Figure 1.

Kaplan-Meier curves for relapse-free survival of Acral and Non-CSD melanoma patients. A, Relapse-free survival time of acral melanoma patients with expression of both NUAK2 and p-Akt(S473) were dramatically shorter than acral melanoma patients with either NUAK2 or p-Akt(S473) alone (P = 0.002). B, Relapse-free survival time of Non-CSD melanoma patients with expression of both NUAK2 and p-Akt(S473) was not different from those of either NUAK2 or p-Akt(S473) alone (P = 0.721). Log rank test was used to detect survival time differences among three groups of patients: 1) Neither NUAK2 nor p-Akt(S473) positive; 2) Either NUAK2 or p-Akt(S473) positive; 3) Both NUAK2 and p-Akt(S473) positive. If we only compare groups 3 and 2 in acral melanomas, the p value is 0.003.
An initial study showed that NUAK2 participates in melanoma cell proliferation by controlling the cell cycle (12). A previous in vivo study using C32 melanoma cells with or without knockdown of NUAK2 showed that the tumor growth of C32 melanoma cells was significantly suppressed by knockdown of NUAK2 (12). In order to elucidate downstream pathways that connect NUAK2 to the cell cycle machinery, we focused on examining the expression of genes in the mTOR, JNK, HIF, cadherin and Rho-Rock pathways using a lentiviral vector containing shRNA targeting NUAK2 (shNUAK2) (Supplementary Fig. S1A). Those analyses revealed that only mTOR is modulated by the knockdown of NUAK2 (12). For further detailed analyses, we used both NUAK2 amplified and PTEN deficient melanoma cells as C32 melanoma cells (NUAK2: 3.94 fold) from Non-CSD melanomas and SM2-1 melanoma cells (NUAK2: 2.12 fold) from acral melanomas, and we used mel18 melanoma cells without amplification of NUAK2 or deficiency of PTEN as a control from acral melanomas (Supplementary Table S2). We initially confirmed that knockdown of NUAK2 in both C32 melanoma cells and SM2-1 melanoma cells reduced cell numbers (Fig. 2A), and the S-phase population was reduced by modulating either the NUAK2 pathway, using shNUAK2, and/or the PI3K pathway, using LY294002 (Fig. 2B). Although the S-phase population of SM2-1 melanoma cells could be examined only by modulating the PI3K pathway using LY294002 due to massive apoptotic destruction of cells by modulating the NUAK2 pathway (Fig. 2C and Supplementary Fig. S4). We then proceeded to examine the expression of genes that control the cell cycle machinery, such as CDK2, CDK4, CDK6, p21 and p27. Knockdown of NUAK2 by shNUAK2 down-regulated the expression of CDK2 and up-regulated the expression of p27. SM2-1 melanoma cells did not express both p21 and p27 (Fig. 3A). Inhibition of the PI3K pathway by LY294002 down-regulated the expression of CDK2 and up-regulated the expression of p21 (Fig. 3B). Immunohistochemical analysis of p21 expression also showed that inhibition of the PI3K pathway by LY294002 increased the percentage of p21-positive cells from 63.7% to 92.3% (Supplementary Fig. S5), and immunohistochemical analysis of the expression of p27 in clinical specimens showed that 86% (6 of 7 cases) of primary melanomas with high-expression of both NUAK2 and p-Akt express p27 (Supplementary Table S3). Knockdown of PTEN by siPTEN slightly increased the expression of CDK2 in mel18 melanoma cells, however the cell number was not affected (Fig. 3C and 3D). And the NUAK2 and PI3K pathways were independently regulated by NUAK2 (Fig. 3E). From these observations, we speculated that both the NUAK2 and PI3K pathways control the expression of CDK2, increase the S-phase population in the cell cycle profile and increase the proliferation of both C32 and SM2-1 melanoma cells (Supplementary Fig. S6). Therefore, we hypothesized that CDK2 might be an efficient target to suppress the proliferation of NUAK2-amplified melanoma cells that are PI3K pathway-activated.
Figure 2.

Both NUAK2 and PI3K pathways regulate the cell cycle machinery. A, Cell proliferation assay showing that knockdown of NUAK2 by shNUAK2 significantly reduces cell proliferation of C32 and SM2-1 melanoma cells (P = 0.0165 and P < 0.0001, respectively) B, FACS analyses showing the S-phase population in the cell cycle profile is reduced by both knockdown of NUAK2 and inhibition of the PI3K pathway in C32 melanoma cells. C, FACS analyses showing the S-phase population in the cell cycle profile is reduced by inhibition of the PI3K pathway in SM2-1 melanoma cells.
Figure 3.

The NUAK2 and PI3K pathways are independently regulated. A, Immunoblots showing the expression of CDK2, CDK4, CDK6, p21 and p27 following the knockdown of NUAK2. Knockdown of NUAK2 decreases the CDK2 expression level and increases the p27 expression level. B, Inhibition of the PI3K pathway by Ly294002 decreases the CDK2 expression level and increases the p21 expression level. C, Immunoblots showing the expression of NUAK2 following the knockdown of PTEN; β-actin was used as a loading control. Knockdown of PTEN by siPTEN did not affect the expression of NUAK2. D, Cell number analysis of mel18 melanoma cells following the knockdown of PTEN. Knockdown of PTEN did not significantly affect cell proliferation in mel18 melanoma cells. E, Immunoblots showing the expression of NUAK2, PTEN, Akt and phospho-Akt following the knockdown of NUAK2; β-actin was used as a loading control. Knockdown of NUAK2 by shNUAK2 slightly increased the expression of Akt but did not affect the expression of p-Akt(S473).
Effects of CDK2 function on NUAK2-amplified melanoma cells
To examine the effect of CDK2 on the proliferation of NUAK2-amplified C32 and SM2-1 melanoma cells, we used siRNA SMARTpools targeting CDK2 (siCDK2), and used mel18 melanoma cells (without aberration of both NUAK2 and PTEN) as a control (Supplementary Table S2). After confirmation of the efficient knockdown by siCDK2 in both of those melanoma cell lines (Supplementary Fig. S7), we evaluated cell numbers at days 2 and 4. Knockdown of CDK2 dramatically reduced the number of C32 and SM2-1 melanoma cells at day 4, whereas knockdown of CDK2 had no effect on the number of mel18 melanoma cells (Fig. 4). Those results suggest that CDK2 preferentially participates in regulating the cell cycle machinery in NUAK2-amplified C32 and SM2-1 melanoma cells. Immunohistochemical analyses also showed a high percentage (35 in 43 cases; 81.40%) of expression of CDK2 in both NUAK2 and p-Akt expressed melanomas (Supplementary Fig. S8 and Table S1). Taken together, we postulated that CDK2 is a candidate target to inhibit the proliferation of NUAK2-amplified melanoma cells.
Figure 4.
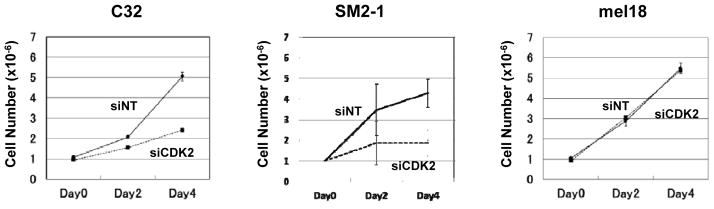
Cell number analysis of C32, SM2-1 and mel18 melanoma cells following the knockdown of CDK2. Knockdown of CDK2 reduced cell proliferation in C32 and SM2-1 melanoma cells (P = 0.003 and P = 0.022, respectively) compared to no reduction in mel18 melanoma cells.
Effect of Roscovitine on melanoma growth
To inhibit CDK2 activity, we evaluated the efficacy of a CDK inhibitor (Roscovitine, also known as Seliciclib and CYC202) that significantly inhibits CDK1 and CDK2 but not CDK4 or CDK6. The proliferation of C32 melanoma cells was inhibited 96.7% (P = 0.011) by 5 μM Roscovitine while only an 18.8% decrease (P = 0.008) occurred in mel18 melanoma cells at the same dose of Roscovitine (Fig. 5A). Similarly, Roscovitine at 5 μM significantly reduced the colony growth of C32 melanoma cells compared to mel18 melanoma cells (Fig. 5B). Cell cycle profile analyses showed that the S-phase population of C32 melanoma cells treated with Roscovitine at 5 μM was significantly reduced from 6.9% to 1.8% (P < 0.001) but 25 μM Roscovitine was required to achieve a similar effect on mel18 melanoma cells (Fig. 5C). In other cell lines with different NUAK2 and PTEN status, Roscovitine had diverse effects depending on the cell lines (Supplementary Fig. S9 and Table S2). We then assessed the effects of Roscovitine on the proliferation of C32, SM2-1 and mel18 melanoma cells in vivo using mice. Tumor growth was significantly suppressed in both C32 melanoma cells (P = 0.0053) and SM2-1 melanoma cells (P = 0.0101) compared to mel18 melanoma cells (P = 0.2136) (Fig. 6). These in vitro and in vivo results indicate that treatment with a low dose of Roscovitine effectively suppresses the growth of NUAK2-amplified melanoma cells.
Figure 5.

A CDK inhibitor targeting CDK2 effectively suppresses the cell proliferation of NUAK2-amplified melanomas. A, Sensitivity of C32 and mel18 melanoma cells to Roscovitine. Short-term cell number analyses showed that the growth of C32 cells is suppressed by Roscovitine at a dose of 5 μM or less. B, Cell proliferation assay showed that the growth of C32 cells is efficiently suppressed by Roscovitine at 5 μM compared to a slight suppression of the growth of mel18 cells at the same dose. C, FACS analysis of the cell cycle profile of C32 and mel18 melanoma cells treated with Roscovitine at 0, 5 and 25 μM. The S-phase population of C32 melanoma cells is significantly reduced by treatment with Roscovitine at 5 μM (upper, n=3, P<0.001), but the S-phase population of mel18 melanoma cells is not changed by treatment of Roscovitine at 5 μM (lower).
Figure 6.
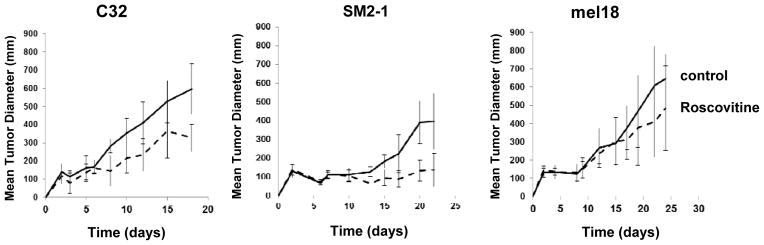
Suppression of tumor growth in mice by Roscovitine. Tumor growth of C32 melanoma cells (left, P = 0.0053) and SM2-1 melanoma cells (middle, P = 0.0101) in mice was suppressed by intraperitoneal administration of Roscovitine, but the tumor growth of mel18 melanoma cells was not suppressed (right, P=0.2136).
Discussion
A wide variety of cancers have genomic aberrations of gains and/or amplification of the long arm of chromosome 1. In melanomas, several candidate genes have been speculated as oncogenes at the long arm of chromosome 1. Further, several cancer related genes have been linked to copy number increases of chromosome 1q, including SETDB1 and MDM4. Analyses of genomic aberrations at 1q in this study using array-CGH data identified a minimal region at 1q32, which is significantly related with tumor thickness in acral melanomas, and show that NUAK2 is a gene that promotes melanomagenesis at this locus. We cannot rule out the possibility that other genes participate in melanomagenesis in association with NUAK2 or alone at 1q32. However, either SETDB1 or MDM4 have quite a low possibility as the responsible gene at 1q32. SETDB1 resides at 1q21.3, where is far away from 1q32. SETDB1 has been identified as a gene related to BRAF mutation (V600E) (25), and a genome-wide association study showed that 1q21.3 is a novel melanoma susceptibility locus in an Australian cohort (26). Those two previous reports suggest that SETDB1 is a gene that participates in melanomagenesis in “Non-CSD melanomas”, not in acral melanomas. The possibility that SETDB1 cooperates with NUAK2 to confer tumorigenicity cannot be excluded, because the long arm of chromosome 1 has two amplification sites of cent-1q21 and 1q32 in melanoma cells. However, the possibility that SETDB1 is an acral melanoma oncogene at 1q32 is quite low. Regarding MDM4, our immunohistochemical studies showed that the possibility of MDM4 as the responsible gene at 1q32 was excluded. Although our initial studies exploring MDM4 as an acral melanoma oncogene from the CGH array data and real-time PCR data revealed the possibility of MDM4 as an oncogene at this locus, our intensive studies ruled out the possibility of MDM4 as an oncogene by clear IHC data since the expression of MDM4 did not correlate with tumor thickness in clinical specimens of acral melanomas. MDM4 has been speculated to participate in melanomagenesis, particularly in Non-CSD melanomas. A previous study of MDM4 expression in melanomas showed almost identical data to our IHC data (27). Thus, MDM4 can be excluded as an acral melanoma oncogene at the 1q32 locus.
Control of the cell cycle machinery has a critical role in regulating cell proliferation and tumor growth of cancer cells. In melanomas, genomic aberrations of melanoma cells have different impacts on the regulation of cell cycle machinery by CDKs, such as that CDK4 is a key driver in NRAS-mutant melanomas and that CDK2 has a pivotal role in melanomas with high expression of MITF (18–20). This study showed that NUAK2 controls CDKs in melanoma cells, and that both the knockdown of NUAK2 and inactivation of the PI3K pathway by Ly294002 suppresses the expression of CDK2. In addition, knockdown of CDK2 using siCDK2 efficiently reduced the cell number. Those results were obtained using three melanoma cell lines (C32, SM2-1 and mel18), in which the genomic status of NUAK2 and PTEN were estimated using real-time PCR and expression levels of either mRNA and protein were estimated by real-time PCR and western blotting, respectively. We selected “Roscovitine” as an inhibitor targeting CDK2 in order to suppress the tumor growth of melanoma cells with NUAK2 amplification. Although Roscovitine has an ability to inhibit several CDKs in addition to CDK2, the results of the cell proliferation assay indicate that Roscovitine can efficiently suppress the proliferation of NUAK2-amplified melanoma cells, and the results of our animal model study are consistent with that.
In summary, this study demonstrates that CDK2 is an effective molecular target for the treatment of NUAK2-amplified melanomas. The inhibition of CDK2 by Roscovitine is a rational approach to reduce cell proliferation and to delay tumor growth, and provides a new therapeutic approach for the treatment of cutaneous melanomas.
Supplementary Material
Acknowledgments
We thank Drs. Murata H and Okuyama R (Shinshu Univ.) for providing the SM2-1 melanoma cell. This work was supported in part by the Intramural Research Program (ZIA BC 010785) of the National Cancer Institute at NIH and Grants-in-Aid for Scientific Research (26221005) from the Japan Society for Promotion of Science.
Footnotes
Conflicts of Interest: The authors have declared that no conflict of interest exists.
References
- 1.Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer. 2012;12(5):349–61. doi: 10.1038/nrc3218. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2(3):210–9. doi: 10.1038/nrc755. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60(7):1800–4. [PubMed] [Google Scholar]
- 6.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8(1):15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 7.Sharpless NE, Chin L. The Ink4a/ARF locus and melanoma. Oncogene. 2003;22:3092–8. doi: 10.1038/sj.onc.1206461. [DOI] [PubMed] [Google Scholar]
- 8.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 9.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485(7399):502–6. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larue L, Dougherty N, Porter S, Mintz B. Spontaneous malignant transformation of melanocytes explanted from Wf/Wf mice with a Kit kinase-domain mutation. Proc Natl Acad Sci USA. 1992;89:7816–20. doi: 10.1073/pnas.89.16.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Namiki T, Tanemura A, Valencia JC, Coelho SG, Passeron T, Kawaguchi M, et al. AMP kinase-related kinase NUAK2 affects tumor growth, migration and clinical outcome of human melanoma. Proc Natl Acad Sci U S A. 2011;108:6597–602. doi: 10.1073/pnas.1007694108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Namiki T, Coelho SG, Hearing VJ. NUAK2: an emerging acral melanoma oncogene. Oncotarget. 2011;2:695–704. doi: 10.18632/oncotarget.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115(4):813–24. doi: 10.1172/JCI24808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ, Powell MB. Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest. 2008;118(11):3660–70. doi: 10.1172/JCI36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007;117(3):719–29. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18(10):1503–10. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du J, Widlund HR, Horstmann MA, Ramaswamy S, Ross K, Huber WE, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell. 2004;6(6):565–76. doi: 10.1016/j.ccr.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Kido K, Sumimoto H, Asada S, Okada SM, Yaguchi T, Kawamura N, et al. Simultaneous suppression of MITF and BRAF V600E enhanced inhibition of melanoma cell proliferation. Cancer Sci. 2009;100(10):1863–9. doi: 10.1111/j.1349-7006.2009.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takata M, Murata H, Saida T. Molecular pathogenesis of malignant melanoma: a different perspective from the studies of melanocytic nevus and acral melanoma. Pigment Cell Melanoma Res. 2010;23(1):64–71. doi: 10.1111/j.1755-148X.2009.00645.x. [DOI] [PubMed] [Google Scholar]
- 22.Curtin JA, Fridlyand J, Kageshita T, Patel MN, Busam KJ, Kutzner HJ, et al. Distinct sets of genetic alterations in melanoma. New Eng J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 23.Watabe H, Valencia JC, Yasumoto K, Kushimoto T, Ando H, Muller J, et al. Regulation of tyrosinase processing and trafficking by organellar pH and by proteasome activity. J Biol Chem. 2004;279:7971–81. doi: 10.1074/jbc.M309714200. [DOI] [PubMed] [Google Scholar]
- 24.Ashida A, Takata M, Murata H, Kido K, Saida T. Pathological activation of KIT in metastatic tumors of acral and mucosal melanomas. Int J Cancer. 2009;124(4):862–8. doi: 10.1002/ijc.24048. [DOI] [PubMed] [Google Scholar]
- 25.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471(7339):513–7. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macgregor S, Montgomery GW, Liu JZ, Zhao ZZ, Henders AK, Stark M, et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21. 3. Nat Genet. 2011;43(11):1114–8. doi: 10.1038/ng.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18:1239–47. doi: 10.1038/nm.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.