

Abstract
In Pd-catalyzed C–N cross-coupling reactions, α-branched secondary amines are difficult coupling partners and the desired products are often produced in low yields. To provide a robust method for accessing N-aryl α-branched tertiary amines, new catalysts have been designed to suppress undesired side reactions often encountered when these amine nucleophiles are used. These advances enabled the arylation of a wide array of sterically encumbered amines, highlighting the importance of rational ligand design in facilitating challenging Pd-catalyzed cross-coupling reactions.
Keywords: C–N cross-coupling, amination, palladium, ligand design, synthetic methods
Tertiary, N-aryl α-branched amines are frequently found as structural components of pharmaceutically relevant compounds and biologically active natural products (Figure 1).[1] Although Pd-catalyzed carbon–nitrogen (C–N) cross-coupling would provide an efficient means of accessing this valuable class of compounds, the use of α-branched secondary amine nucleophiles has seen only limited success and in many instances low yields of the desired product are obtained.[2] Other methods for preparing tertiary N-aryl α-branched amines rely on the addition of an amine to an aryne[3] or nucleophilic aromatic substitution.[4] While effective, these methods typically have a narrow substrate scope or result in a mixture of regioisomeric products.[3] Copper-catalyzed electrophilic amination has also been utilized,[5] with a recent report by Lalic demonstrating its effectiveness for the arylation of sterically hindered secondary O-benzoyl hydroxylamine electrophiles.[5b] Despite these advances, there remains no general method for the direct arylation of α-branched secondary amines. Therefore, we sought to develop a catalyst system capable of cross-coupling sterically encumbered secondary amines.
Figure 1.
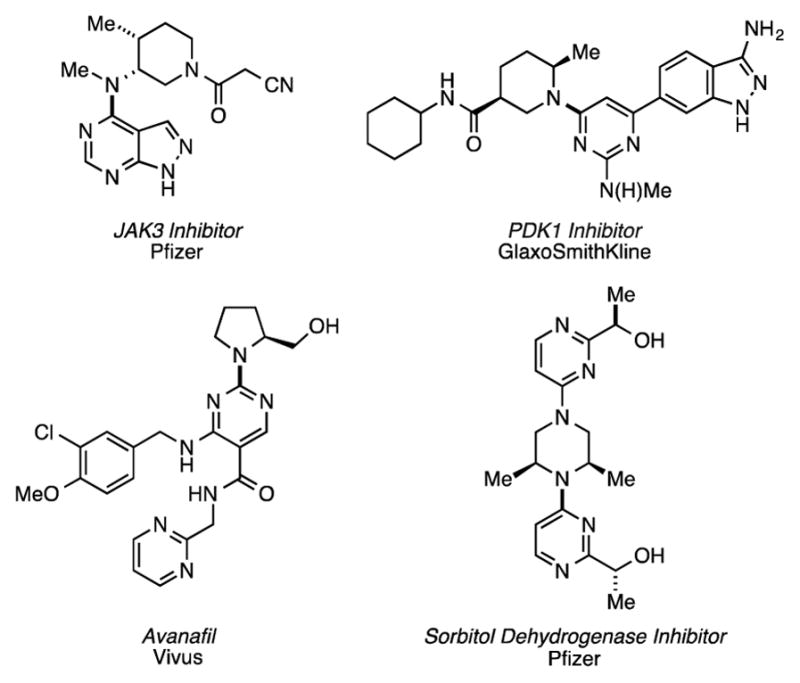
Selected examples of biologically active compounds containing tertiary N-aryl α-branched amines.[1]
The development of a highly effective catalyst system for the arylation of α-branched secondary amines must address the specific challenges presented by these coupling partners. Their poor nucleophilicity as a consequence steric hindrance can lead to slower rates of amine transmetalation, resulting in the competitive reaction of the alkoxide base and formation of the corresponding aryl tert-butyl ether (ArOtBu) (V, Figure 2). Additionally, β-hydride elimination may occur from the intermediate Pd(II)-amido complex[6,7] (IV, Figure 2) leading to the formation of the reduced arene (VI, Figure 2). In this regard, the supporting ligand for the palladium catalyst must be carefully designed in order to facilitate the preferential formation of the desired aryl amine while suppressing side reactions.
Figure 2.
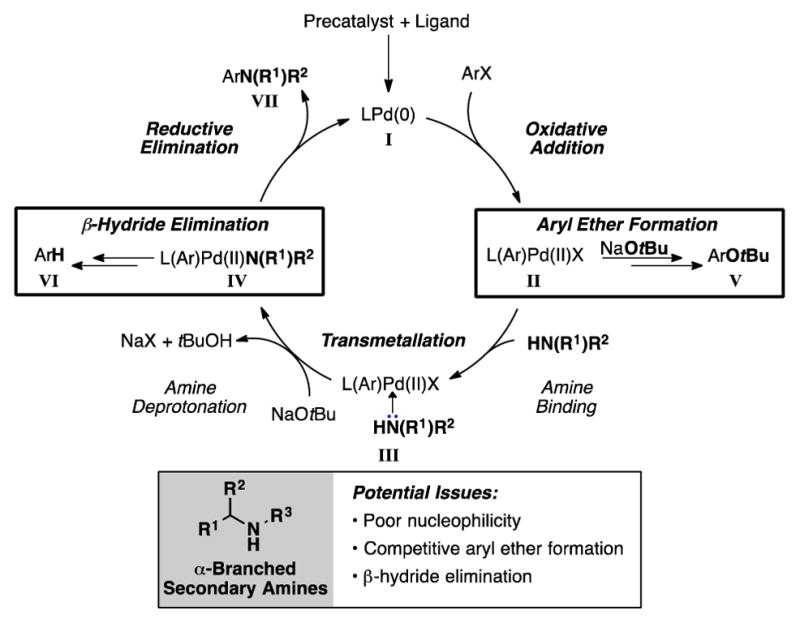
Proposed catalytic cycle and potential challenges presented by sterically hindered α-branched secondary amine nucleophiles.
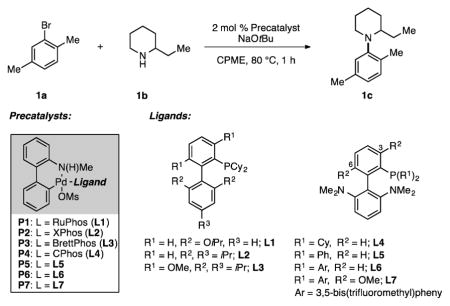
We began our investigation by examining the effect of the supporting ligands on the efficiency of the catalyst system for the reaction shown in Table 1.[8] RuPhos(L1)-based catalyst systems have been demonstrated to be highly effective for the cross-coupling of secondary amines,[9] including some cases of reactions between sterically demanding coupling partners.[2a,2c] However, when RuPhos precatalyst P1 was used in the reaction of 2-bromo-p-xylene (1a) and 2-ethylpiperidine (1b) only a 10% yield of the desired product was obtained (Table 1, entry 1). Other biaryl phosphine ligands such as XPhos (L2) and BrettPhos (L3) have also been used for promoting Pd-catalyzed C–N bond formation.[9] Nevertheless, these catalyst systems (P2 and P3, respectively) proved to be inefficient in facilitating the desired transformation (Table 1, entries 2–3). In all cases, the major byproduct was the reduced arene, which presumably arises as a result of β-hydride elimination.[10]
Table 1.
Supporting Ligand Evaluation.[a]
| ||||
|---|---|---|---|---|
| Entry | Precatalyst | Conversion | Reduction | Yield |
| 1 | P1 | 100% | 68% | 10% |
| 2 | P2 | 100% | 85% | 15% |
| 3 | P3 | 37% | 15% | 2% |
| 4 | P4 | 100% | 53% | 27% |
| 5 | P5 | 100% | 18% | 77% |
| 6 | P6 | 94% | Trace | 85% |
| 7 | P7 | 100% | Trace | 93%[b],[c] |
Reaction conditions: 1a (0.25 mmol), 1b (0.30 mmol), NaOtBu (0.35 mmol), 2 mol % precatalyst, CPME (0.5 mL), 80 °C, 1 h. Conversion, C–N cross-coupling, and reduction product yields were measured by GC analysis of the crude reaction mixture using dodecane as the internal standard.
The reaction also produced 6% of the corresponding ArOtBu.
Isolated yield: 89% (1 mmol scale, average of two runs).
CPME = cyclopentyl methyl ether.
Given these results, we turned to CPhos (L4, Table 1), which has been demonstrated to suppress β-hydride elimination in Pd-catalyzed Negishi cross-coupling reactions.[11] Indeed, CPhos precatalyst P4 produced aryl amine 1c in improved yield, although the reduced arene remained the major product (Table 1, entry 4).
In the proposed catalytic cycle, the β-hydride elimination pathway competes with reductive elimination from the Pd(II)-amido intermediate (IV, Figure 2). We thus envisoned that using a less electron-rich biaryl phosphine ligand would increase the rate of C–N reductive elimination.[12] A less electron-rich biaryl phosphine ligand could also increase the rate of transmetalation (amine binding and deprotonation, Figure 2) by rendering the Pd(II) intermediates II and III more electrophilic (Figure 2).[13] Based on this hypothesis, we examined a catalyst system utilizing the ligand L5 (P5, Table 1).[14,15] The use of precatalyst P5 dramatically increased the yield of 1c along while decreasing the amount of reduced arene formed (Table 1, entry 5). Following these results, we changed the phosphorus substituents from phenyl to 3,5-bis(trifluoromethyl)phenyl groups to provide ligand L6 (P6, Table 1); this led to additional improvement in the yield and further diminished the formation of the reduced arene (Table 1, entry 6). To achieve additional improvements in catalyst performance, we incorporated methoxy groups in the 3 and 6 positions of the biaryl framework (Table 1) as these groups are known to increase the rate of reductive elimination from Pd(II) complexes.[16] This modification produced L7 (P7), which provided the most efficient catalyst system for the transformation (Table 1, entry 7).[17]
Precatalyst P7 was found to enable a wide variety of C–N cross-coupling reactions with α-branched secondary amines (Scheme 1). Hindered cyclic secondary amines were found to be well-tolerated, including in reactions with aryl halides containing ortho-substituents (2a, 2c, 2e, 2g, and 2i, Scheme 1). Lower yields were obtained in the more sterically encumbered cases,[18] where formation of the reduced arene byproduct was observed. Acyclic α-branched amines could also be efficiently arylated (2b and 2h, Scheme 1). Previously, the arylation of diisopropylamine via Pd-catalyzed C–N cross-coupling has resulted in very low yields,[2f,20] presumably due to its steric hindrance. By using P7, however, diisopropylamine was successfully arylated in 65% yield (2h, Scheme 1), although additional equivalents of amine and base were necessary to favor formation of the desired product.[21,22]
Scheme 1.

Scope of C–N cross-coupling reactions using P7. Reaction conditions: aryl halide (1.0 mmol), amine (1.2 mmol), NaOtBu (1.4 mmol), 2 mol % P7, 0–2 mol % L7, CPME (2 mL), 60–80 °C, 6–16 h. Yields are of isolated products, average of two runs. [a] 1:49 cis:trans isomers of the arylated amine. Determined by GC analysis of the crude reaction mixture. 2% reduction, 4% ArOtBu. [b] 9% ArOtBu. [c] 27% reduction, 6% ArOtBu [d] 22:1 cis:trans isomers of the arylated amine. Determined by GC analysis of the crude reaction mixture. [e] 28% reduction. [f] K3PO4 (6.0 mmol) used as base. [g] 34% reduction. [h] Amine (9.6 mmol), NaOtBu (10.8 mmol), 7% reduction, 9% ArOtBu. [i] 37% reduction.
We were interested in applying the developed conditions to the amination of heteroaryl halides due to their presence in many pharmaceutically relevant compounds.[2] However, our initial attempts to utilize activated heteroaryl electrophiles (3a, 3b, and 3c, Scheme 2) resulted in low yields and the formation of significant amounts of the corresponding ArOtBu.[23,24] Through systematic ligand modification[25] we found that ligand L8 (P8, Scheme 2) provided higher yields in these cases. With all other substrates, P7 was again very effective in producing high yields of the desired product. In certain instances, the use of additional equivalents of the amine was necessary to further deter the formation of the ArOtBu (3a, 3g, and 3i, Scheme 2). Additionally, a trace of the epimerized product was observed in cases where cis-2,6-dimethylpiperidine (3g, Scheme 2) or an enantiomerically enriched amine was used (3h and 3i, Scheme 2). Despite these considerations, the combined substrate scope using precatalysts P7 and P8 allows for efficient cross-coupling of a wide variety of challenging α-branched secondary amines with different heteroaryl halides (Scheme 2).
Scheme 2.

The scope of C–N cross-coupling reactions with heteroaryl halides and hindered secondary amines. Reaction conditions: aryl halide (1.0 mmol), amine (1.2 mmol), NaOtBu (1.4 mmol), 2–3 mol % P7 or P8, 0–2 mol % L7 (used only with P7), CPME (2 mL), 60–80 °C, 16 h. Yields are of isolated products, average of two runs. [a] Amine (2.4 mmol), NaOtBu (2.8 mmol). [b] 9% reduction, 8% ArOtBu. [c] 2% reduction, 3% ArOtBu. [d] 13% reduction. [e] Amine (3.6 mmol), NaOtBu (4.2 mmol); 20:1 cis:trans isomers of the arylated amine product. Determined by GC analysis of the crude reaction mixture. [f] Starting amine ee: 99% ee; Product ee: 98% ee. [g] Amine (2.4 mmol), NaOtBu (2.8 mmol), dioxane (2 mL); 24% ArOtBu, 6% reduction; Starting amine ee: ≥97% ee; Product ee: 83% ee.
In summary, we have developed two new catalyst systems for the arylation of sterically demanding α-branched secondary amines. Notably, the unprecedented levels of reactivity in C–N cross-coupling reactions with these amines are achieved due to the ability of the new precatalysts to suppress both the β-hydride elimination pathway and arylation of the alkoxide base. Overall, this work highlights the potential of rational ligand design to modulate catalyst behavior and ultimately facilitate the cross-coupling of sterically demanding amine coupling partners.
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health for support of this work (grant GM58160). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health. N.H.P. acknowledges funding from a National Science Foundation Graduate Research Fellowship. We thank Dr. Xiaohua Huang (MIT) for previous work on the arylation of diisopropylamine. We thank Dr. Michael T. Pirnot (MIT), Dr. Aaron C. Sather (MIT), and Dr. Christine Nguyen (MIT) for aid in the preparation of this manuscript. MIT holds or has filed patents on the ligands and precatalysts used in this work for which S.L.B and current and/or former coworkers receive royalty payments.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) DeGoey DA, Randolph JT, Liu D, Pratt J, Hutchins C, Donner P, Krueger AC, Matulenko M, Patel S, Motter CE, Nelson L, Keddy R, Tufano M, Caspi DD, Krishnan P, Mistry N, Koev G, Reisch TJ, Mondal R, Pilot-Matias T, Gao Y, Beno DW, Maring CJ, Molla A, Dumas E, Campbell A, Williams L, Collins C, Wagner R, Kati WM. J Med Chem. 2014;57:2047. doi: 10.1021/jm401398x. [DOI] [PubMed] [Google Scholar]; b) Medina JR, Becker CJ, Blackledge CW, Duquenne C, Feng Y, Grant SW, Heerding D, Li WH, Miller WH, Romeril SP, Scherzer D, Shu A, Bobko MA, Chadderton AR, Dumble M, Gardiner CM, Gilbert S, Liu Q, Rabindran SK, Sudakin V, Xiang H, Brady PG, Campobasso N, Ward P, Axten JM. J Med Chem. 2011;54:1871. doi: 10.1021/jm101527u. [DOI] [PubMed] [Google Scholar]; c) Bringmann G, Gulder T, Hertlein B, Hemberger Y, Meyer F. J Am Chem Soc. 2010;132:1151. doi: 10.1021/ja9097687. [DOI] [PubMed] [Google Scholar]; d) Yamada K, Matsuki K, Omori K, Kikkawa K. 6,797,707. US Patent. 2004; e) Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, Rizzuti BJ, Sawyer PS, Perry BD, Brissette WH, McCurdy SP, Kudlacz EM, Conklyn MJ, Elliott EA, Koslov ER, Fisher MB, Strelevitz TJ, Yoon K, Whipple DA, Sun J, Munchhof MJ, Doty JL, Casavant JM, Blumenkopf TA, Hines M, Brown MF, Lillie BM, Subramanyam C, Shang-Poa C, Milici AJ, Beckius GE, Moyer JD, Su C, Woodworth TG, Gaweco AS, Beals CR, Littman BH, Fisher DA, Smith JF, Zagouras P, Magna HA, Saltarelli MJ, Johnson KS, Nelms LF, Des Etages SG, Hayes LS, Kawabata TT, Finco-Kent D, Baker DL, Larson M, Si MS, Paniagua R, Higgins J, Holm B, Reitz B, Zhou YJ, Morris RE, O’Shea JJ, Borie DC. Science. 2003;302:875. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]; f) Chu-Moyer MY, Ballinger WE, Beebe DA, Berger R, Coutcher JB, Day WW, Li J, Mylari BL, Oates PJ, Weekly RM. J Med Chem. 2001;45:511. doi: 10.1021/jm010440g. [DOI] [PubMed] [Google Scholar]
- 2.For selected references containing examples of Pd-catalyzed C–N bond formation with α-branched secondary amines, see: Bourbeau MP, Ashton KS, Yan J, StJean DJ., Jr J Org Chem. 2014;79:3684. doi: 10.1021/jo500336e.Samblanet DC, Schmidt JAR. J Organomet Chem. 2012;720:7.Foo K, Newhouse T, Mori I, Takayama H, Baran PS. Angew Chem. 2011;123:2768. doi: 10.1002/anie.201008048.Foo K, Newhouse T, Mori I, Takayama H, Baran PS. Angew Chem, Int Ed. 2011;50:2716. doi: 10.1002/anie.201008048.Organ MG, Abdel-Hadi M, Avola S, Dubovyk I, Hadei N, Kantchev EA, O’Brien CJ, Sayah M, Valente C. Chem Eur J. 2008;14:2443. doi: 10.1002/chem.200701621.Peng ZH, Journet M, Humphrey G. Org Lett. 2006;8:395. doi: 10.1021/ol052578p.Iyer S, Kulkarni GM, Ramesh C, Satttar AK. Indian J Chem, Sec B. 2005;44B:1894.Saroja G, Pingzhu Z, Ernsting NP, Liebscher J. J Org Chem. 2004;69:987. doi: 10.1021/jo035204n.van Delden RA, Hurenkamp JH, Feringa BL. Chem Eur J. 2003;9:2845. doi: 10.1002/chem.200204660.Qadir M, Priestley RE, Rising TWDF, Gelbrich T, Coles SJ, Hursthouse MB, Sheldrake PW, Whittall N, Hii KK. Tetrahedron Lett. 2003;44:3675.Tanatani A, Mio MJ, Moore JS. J Am Chem Soc. 2001;123:1792. doi: 10.1021/ja003678n.Wagaw S, Rennels RA, Buchwald SL. J Am Chem Soc. 1997;119:8451.Marcoux JF, Wagaw S, Buchwald SL. J Org Chem. 1997;62:1568.Zhao SH, Miller AK, Berger J, Flippin LA. Tetrahedron Lett. 1996;37:4463.
- 3.a) Bolliger J, Frech C. Tetrahedron. 2009;65:1180. [Google Scholar]; b) Lin W, Sapountzis I, Knochel P. Angew Chem. 2005;117:4330. doi: 10.1002/anie.200500443. [DOI] [PubMed] [Google Scholar]; Lin W, Sapountzis I, Knochel P. Angew Chem, Int Ed. 2005;44:4258. doi: 10.1002/anie.200500443. [DOI] [PubMed] [Google Scholar]; c) Shi L, Wang M, Fan CA, Zhang FM, Tu YQ. Org Lett. 2003;5:3515. doi: 10.1021/ol0353868. [DOI] [PubMed] [Google Scholar]; d) Tripathy S, LeBlanc R, Durst T. Org Lett. 1999;1:1973. [Google Scholar]; e) Walters MA, Shay JJ. Tetrahedron Lett. 1995;36:7575. [Google Scholar]; f) Bhaskar Kanth JV, Periasamy M. J Org Chem. 1993;58:3156. [Google Scholar]; g) Wickham PP, Hazen KH, Guo H, Jones G, Reuter KH, Scott WJ. J Org Chem. 1991;56:2045. [Google Scholar]; h) Biehl ER, Razzuk A, Jovanovic MV, Khanapure SP. J Org Chem. 1986;51:5157. [Google Scholar]; i) Huisgen R, Zirngibl L. Chem Ber. 1958;91:2375. [Google Scholar]
- 4.a) Waldvogel S, Faust A, Wolff O. Synthesis. 2009:155. [Google Scholar]; b) Nijhuis WHN, Verboom W, Abu El-Fadl A, Harkema S, Reinhoudt DN. J Org Chem. 1989;54:199. [Google Scholar]
- 5.a) Qian X, Yu Z, Auffrant A, Gosmini C. Chem Eur J. 2013;19:6225. doi: 10.1002/chem.201300229. [DOI] [PubMed] [Google Scholar]; b) Rucker RP, Whittaker AM, Dang H, Lalic G. Angew Chem. 2012;124:4019. doi: 10.1002/anie.201200480. [DOI] [PubMed] [Google Scholar]; Rucker RP, Whittaker AM, Dang H, Lalic G. Angew Chem, Int Ed. 2012;51:3953. doi: 10.1002/anie.201200480. [DOI] [PubMed] [Google Scholar]; c) Barker TJ, Jarvo ER. J Am Chem Soc. 2009;131:15598. doi: 10.1021/ja907038b. [DOI] [PubMed] [Google Scholar]; d) Berman AM, Johnson JS. J Org Chem. 2005;71:219. doi: 10.1021/jo051999h. [DOI] [PubMed] [Google Scholar]
- 6.a) Beletskaya IP, Bessmertnykh AG, Guilard R. Tetrahedron Lett. 1999;40:6393. [Google Scholar]; b) Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805. [Google Scholar]; c) Hartwig JF, Richards S, Barañano D, Paul F. J Am Chem Soc. 1996;118:3626. [Google Scholar]
- 7.Reduction of the aryl halide can occur during catalyst activation from Pd(II) salts (see ref. 6c). However, this is unlikely with the N-methyl 2-aminobiphenyl palladium methanesulfonate precatalysts used in this study due to their mechanism of activation, see ref. 8a
- 8.The presence of carbazole from the activation of the 2-amino biphenyl palladium methanesulfonate precatalysts inhibited the reaction. Therefore, the N-methyl 2-aminobiphenyl palladium methanesulfonate precatalysts were used instead. Bruno NC, Niljianskul N, Buchwald SL. J Org Chem. 2014;79:4161. doi: 10.1021/jo500355k.Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916. doi: 10.1039/C2SC20903A.
- 9.a) Maiti D, Fors BP, Henderson JL, Nakamura Y, Buchwald SL. Chem Sci. 2011;2:57. doi: 10.1039/C0SC00330A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Surry DS, Buchwald SL. Chem Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Experiments on a related substrate using a deuterated amine nucleophile have shown that the reduced arene product does arise from a β-hydride elimination process.
- 11.a) Yang Y, Niedermann K, Han C, Buchwald SL. Org Lett. 2014;16:4638. doi: 10.1021/ol502230p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Han C, Buchwald SL. J Am Chem Soc. 2009;131:7532. doi: 10.1021/ja902046m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phosphine ligands containing aryl rings with electron-withdrawing substituents have been shown to facilitate a faster rate of C–N reductive elimination from a Pd(II) amido complex, see: Hartwig JF. Inorg Chem. 2007;46:1936. doi: 10.1021/ic061926w.
- 13.a) Hicks JD, Hyde AM, Cuezva AM, Buchwald SL. J Am Chem Soc. 2009;131:16720. doi: 10.1021/ja9044357. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sergeev AG, Artamkina GA, Beletskaya IP. Tetrahedron Lett. 2003;44:4719. [Google Scholar]
- 14.DeBergh JR, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:10638. doi: 10.1021/ja405949a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Changing the substituents on the phosphorus from cyclohexyl to aryl groups can also reduce the size of the ligand, which could potentially make the catalyst more accommodating to larger amine nucleophiles.
- 16.For examples of the effect of incorporating methoxy groups on the ligand biaryl structure in Pd-catalyzed carbon–heteroatom bond forming reactions, see refs. 9b, 13a, and: Wu X, Fors BP, Buchwald SL. Angew Chem. 2011;123:10117. doi: 10.1002/anie.201104361.X Wu, Fors BP, Buchwald SL. Angew Chem, Int Ed. 2011;50:9943. doi: 10.1002/anie.201104361.
- 17.In this case, the difference in performance between ligands L6 and L7 was not significant. However, L7 was found to perform considerably better than L6 in the reaction with other substrates, particularly aryl chlorides, see Supporting Information.
- 18.The C–F bond length is more similar to the C–O bond length than C–H, making the steric effects of an ortho-fluoro substituent slightly more significant than an ortho-hydrogen substituent, see: K. Müller, C. Faeh, F. Diederich, Science 2007, 317, 1881.
- 19.The amounts of the corresponding reduced arene and ArOtBu byproducts observed have been indicated in the table footnotes. In most cases, these byproducts could be readily separated from the desired aryl amine product. Only in the case of 2c was the separation difficult and the isolated material contained <5% of the corresponding ArOtBu.
- 20.The cross-coupling of diisopropylamine with 4-bromoanisole was reported by Herrmann to provide the aryl amine product in 78% yield. In our hands, the products under these conditions resulted from the arylation of the corresponding N-isopropylpropan-2-imine see: Herrmann WA, Böhm WVP, Reisinger C-P. J Organomet Chem. 1999;576:23. and the Supporting Information.
- 21.A control experiment produced none of the arylated diisopropylamine or the corresponding ArOtBu, see Supporting Information.
- 22.It was found that having an excess of amine relative to NaOtBu lead to incomplete conversion of the aryl electrophile. As such, the same amine to base ratio was maintained for all reactions, see Supporting Information.
- 23.Control experiments for substrates 3a, 3b, and 3c showed no formation of the product or the corresponding ArOtBu, see Supporting Information.
- 24.When P7 is used, the yields of 3a, 3b and 3c are 5%, 60%, and 70% respectively, see Supporting Information.
- 25.See Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.