

Abstract
Uveal melanoma (UM) patients with metastatic disease usually die within one year, emphasizing an urgent need to develop new treatment strategies for this cancer. MEK inhibitors improve survival in cutaneous melanoma patients but show only modest efficacy in metastatic UM patients. In this study, we screened for growth factors that elicited resistance in newly characterized metastatic UM cell lines to clinical grade MEK inhibitors, trametinib and selumetinib. We show that neuregulin 1 (NRG1) and hepatocyte growth factor (HGF) provide resistance to MEK inhibition. Mechanistically, trametinib enhances the responsiveness to NRG1, and sustained HGF mediated activation of AKT. Individually targeting ERBB3 and cMET, the receptors for NRG1 and HGF respectively, overcomes resistance to trametinib provided by these growth factors and by conditioned medium from fibroblasts that produce NRG1 and HGF. Inhibition of AKT also effectively reverses the protective effect of NRG1 and HGF in trametinib-treated cells. UM xenografts growing in the liver in vivo and a subset of liver metastases of UM patients express activated forms of ERBB2 (the co-receptor for ERBB3) and cMET. Together, these results provide preclinical evidence for the use of MEK inhibitors in combination with clinical-grade anti-ERBB3 or anti-cMET monoclonal antibodies in metastatic UM.
Introduction
Uveal melanoma (UM) originates from the melanocytes within the iris, choroid and ciliary body (1). Each year, approximately 2,500 new patients will be diagnosed with this disease in the United States. Half of these patients will develop metastases, typically in the liver, within fifteen years of initial diagnosis with a peak of metastasis between 2 and 5 years. Although there are effective therapeutic strategies to prevent local recurrence and to eradicate primary UM, patients with metastatic disease are found to be refractory to current chemotherapies and immune checkpoint blockers and usually die within a year (2).
Recent advances have identified genetic alterations in UM. In contrast to its cutaneous counterpart, oncogenic BRAF mutations are infrequent in UM (3–6). Activating mutations in two alpha subunits of the heterotrimeric G proteins, GNAQ and GNA11, are found in 80% of UMs in mutually exclusive manner and are believed to occur at an early stage of disease (7–11). The GNAQ and GNA11 mutations are typically in Q209 but less frequently in R183. Other studies have also identified recurrent mutations in SF3B1 (12–14), a RNA splicing factor, and EIF1AX (12) in primary UM with disomy 3 and associate with low metastatic potential. Inactivating mutations in the tumor suppressor BRCA1 associated protein 1 (BAP1) on chromosome 3 are found in 32–50% of primary UM and associate with a more aggressive/higher likelihood of metastasis (15–17).
Oncogenic mutations in GNAQ and GNA11 abrogate their intrinsic GTPase activities, resulting in activation of the RAF/MEK/ERK1/2 and protein kinase C (PKC) signaling, JNK and p38 via regulation of the small GTPases of RhoA and Rac1 (18). These signaling pathways promote tumor proliferation and growth. Knockdown of GNAQ in mutant but not wild type UM cell lines diminishes ERK1/2 activation, induces cell cycle arrest (8, 19) and AMP-activated protein kinase-dependent autophagic cell death (20). While these findings emphasize the potential of targeted therapy in UM, directly targeting mutant GNAQ and GNA11 has proved to be structurally challenging.
Targeting MEK with small molecule inhibitors such as trametinib (GSK1120212) and selumetinib (AZD6244) has been pursued in clinical trials for melanoma. Trametinib monotherapy has achieved 25–40% partial/complete response rates in BRAF V600E/K cutaneous melanoma patients (21). By contrast, while trametinib is recently FDA-approved for cutaneous melanoma, it is largely ineffective in uveal tumors. In a phase I trial containing 16 UM patients, 8 patients had stable disease but no partial or complete responses were observed (21). In a phase II trial, selumetinib improved progression free survival compared to standard chemotherapy (15.9 vs 7.0 weeks) (22). Although overall survival was improved with selumetinib, the improvement did not reach statistical significance, possibly due to the cross-over study design. Thus, targeting MEK alone in UM patients has limited clinical benefit. In UM cells line studies, MEK inhibition alone elicited a cell cycle arrest but did not induce apoptosis (19). To determine the underlying mechanisms, we explored the adaptive and/or innate resistance pathways that bypass the requirement for MEK/ERK1/2 signaling in UM.
In this work, we show that two growth factors, NRG1 and HGF, mediate resistance to the MEK inhibitors trametinib (23) and selumetinib (24) in metastatic human UM cells. Mechanistically, MEK inhibition enhances responsiveness to NRG1 and promotes sustained HGF-induced phosphorylation of cMET. Targeting NRG1-ERBB3 or HGF-cMET signaling overcomes the respective growth factor-mediated resistance. Also, fibroblast-derived factors act in a paracrine manner to induce resistance to trametinib through activating NRG1-ERBB3 and HGF-cMET signaling in UM cells. Lastly, ERBB2 (the co-receptor for ERBB3) and cMET were activated in an orthotopic metastatic UM mouse model and in a subset of liver metastases of UM. Together, these data suggest that co-targeting MEK with ERBB3 and/or cMET may enhance the efficacy of MEK inhibitor in advanced stage UM patients.
Materials and Methods
Metastatic UM cell lines and cell culture
UM001, UM003 and UM004 were derived from liver, retroperitoneal and orbital metastases of human UM, respectively. The mutational status of UM001 and UM003 cells were described previously (25). Mutational analysis of GNAQ was performed by Sanger sequencing. UM001 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 10% non-essential amino acids, 2 mM L-glutamine, 10 mM Hepes buffer, 50 IU/ml penicillin and 50 mg/ml streptomycin. UM003 cells and UM004 cells were maintained in MEM medium containing 15% (UM003 cells) or 10% (UM004 cells) heat-inactivated FBS and penicillin-streptomycin. Human telomerase reverse transcriptase (hTERT) immortalized foreskin fibroblastic BJ1 cells were provided by Dr. Ubaldo Martinez-Outschoorn (Thomas Jefferson University, PA). The human embryonic lung fibroblastic Wi38 cell line was purchased from ATCC (Manassas, VA). HT-BJ1 and Wi38 cell lines were maintained in DMEM medium supplemented with 10% heat-inactivated FBS and penicillin-streptomycin. To collect conditioned medium from HT-BJ1 and Wi-38, cells were cultured in UM001 or UM003/4 medium for three days.
Inhibitors, growth factors and function-blocking antibodies
Trametinib (GSK1120212), selumetinib (AZD6244), lapatinib, crizotinib and MK2206 were purchased from Selleck Chemicals (Houston, TX). Recombinant human NRG1, PDGF-BB, and IGF-1 were purchased from Cell Signaling Technology (Beverly, MA); recombinant human EGF was purchased from Lonza Walkersville Inc141 (Walkersville, MD); recombinant human HGF was provided by Michael P. Lisanti (University of Manchester, UK). Humanized ERBB3 monoclonal antibody U3-1287 was provided by U3 Pharma (Martinsried, Germany).
Short-interfering RNA (siRNA) and transfection
UM001 and UM004 cells (3×105) were seeded in 6-well plates. The next day, cells were transfected for 4–6 hours with chemically synthesized siRNAs at a final concentration of 25 nM using Lipofectamine™ RNAiMAX (Invitrogen, Carlsbad, CA) as previously described (26). cMET siRNAs (#1: GAAGAUCAGUUUCCUAAUU; #2: CCAGAGACAUGUAUGAUAA) and ERBB2 siRNAs (#1: GGACGAAUUCUGCACAAUG; #2: GACGAAUUCUGCACAAUGG) were purchased from Dharmacon Inc. (Lafayette, CO). The non-targeting siRNA (UAGCGACUAAACACAUCAAUU) was used as a control.
MTS Assay
UM001, UM003 and UM004 cells (3–6 ×103 cells per well) were plated overnight into 96-well plates and treated with trametinib (0.003–0.4 uM) or vehicle (DMSO) for 72 hours. Each assay was performed in triplicate. Cell growth inhibition was assayed by MTS (Promega Corporation, Madison, WI) according to manufacturer's instruction (error bars reflect +/− SEM of 3 independent experiments). IC50 and IC25 values were calculated using Graphpad Prism.
Western blotting
Cells were washed in cold PBS and lysed directly in Laemmli sample buffer. Lysates were resolved by SDS polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were blocked with 1% BSA and incubated with indicated primary antibodies overnight at 4°C. Western assays were detected using the horseradish peroxidase-conjugated secondary antibodies followed by development using chemiluminescence substrate (Pierce, Rockford, IL). Primary antibodies used were: ERK2 (D-2) from Santa Cruz Biotech. Inc. (Santa Cruz, CA); ERBB3 (1B2E), phospho-ERBB3 Y1197 (C56E4), phospho-ERBB3 Y1289 (21D3), ERBB2 (D8F12), phospho-ERBB2 Y1196 (D66B7), MET, phospho-MET Y1234/1235 (D26), PDGFR, phospho-PDGFRβ Y751 (C63G6), EGFR, phospho-EGFR Y845, IGF1R, phospho-IGF1Rβ Y1131, AKT, phospho-AKT T308 (C31E5E), phospho-AKT S473 (D9E) and phospho-ERK1/2 (D13.14.4E), and phospho-TSC2 T1462 from Cell Signaling Tech. (Danvers, MA); Actin from Sigma-Aldrich (St Louis, MO). Chemiluminescence was visualized on a Versadoc MultiImager and quantitated using Quantity-One software (Bio-Rad, Hercules, CA).
Flow cytometry
Cells were trypsinized, washed with cold PBS and resuspended in 0.5 ml cold PBS to achieve single cell suspension. Cells were then fixed in 4.5 ml 70% ethanol for 2 hours, followed by centrifugation at 800 × g for 5 minutes. Cell pellets were washed with cold PBS and resuspended in 1 ml 0.1% (v/v) Triton X-100 staining solution containing 100 ng/ml RNase and 40 μg/ml propidium iodide (PI) for 30 minutes at room temperature. Staining was then analyzed by flow cytometry on a BD FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ). Data was analyzed by FlowJo software (Tree Star, Inc., Ashland, OR). To determine cell surface expression of ERBB3 and cMET, UM cells were incubated in PBS with 2% BSA and 50 μl PE-conjugated anti-ERBB3 antibody (R&D Systems, Minneapolis, MN), PE-conjugated anti-cMET antibody or isotype control IgG antibody on ice for 45 min. Washed cells were analyzed by flow cytometry and data were analyzed by FlowJo software.
Cell viability assays
Cells were plated at a confluency of 3×105 per well in 6-well plates. The next day, growth factors, drugs or function-blocking antibodies were added as indicated. Cells were cultured for additional three days (UM001 cells) or five days (UM003 cells, for which medium and additives were replenished once), at which time AlamarBlue® (Invitrogen, Grand Island, NY) was added to each well and allowed to reduce for approximately 1 hour. 120 μl of medium was collected in triplicate from each condition and absorbance readings for oxidized and reduced AlamarBlue® were taken at wavelengths 600nM and 570nM, respectively, in a Multiskan® Spectrum spectrophotometer (Thermo Scientific, Waltham, MA). The change in viability was calculated from the resulting absorbance values using the manufacturer's guidelines. All conditions were normalized to the DMSO control.
Cell growth assays
Cells were plated as for viability assays and stained with crystal violet solution (1% crystal violet, 10% buffered formalin) for 30 minutes. After decanting the staining solution, wells were thoroughly washed in distilled water and air dried. Plates were imaged by scanning while colonies were imaged on a Nikon™ Eclipse Ti inverted microscope (Nikon, Tokyo, Japan) with NIS-Elements AR 3.00 software (Nikon, Tokyo, Japan).
Immunohistochemistry
UM001 cells (1 × 106) were injected into the liver of NSG mice or hHGFki mice (STOCK Hgftm1.1(HGF)Aveo Prkdcscid/J, Jackson Labs) and allowed to colonize for 4–5 weeks (NSG mice) or 8 weeks (hHGFki mice). Tissue samples from UM001 xenografts were fixed in formalin overnight. Paraffin-embedded tissue sections were deparaffinized and antigen retrieval was accomplished using high pH conditional buffer. Sections were incubated with anti-phospho-ERBB2 Y1221/Y1222 (6B12) and anti-phospho-cMET Y1234/Y1235 (D26) antibodies (Cell Signaling Technology) overnight. The next day, sections were incubated for 30 minutes in ImmPRESS UNIVERSAL Reagent (Vector Laboratories), following by incubating for 2–5 minutes in ImmPACT NOVA-RED (Vector Laboratories). Sections were counterstained with hematoxylin.
Expression of phospho-ERBB2 and phospho-cMET in the liver biopsy specimens from human UM patients was carried out in a Ventana Ultra stainer (Ventana Medical Systems). In brief, heat-induced epitope retrieval was performed using cell conditioner I buffer. Sections were then incubated with pre-diluted primary antibodies, following by Alkaline Phosphatase Multimer (Ventana Medical Systems) incubation. The fast red chromogen was applied for bright fuchsia color development. The sections were counter-stained with hematoxylin for microscopic evaluation. The histological evaluation of individual tumor specimens was carried out with slides stained with hematoxylin and eosin (H&E). The intensity of staining and the percentage of positive cells were semi-quantatively evaluated by a board certified pathologist (Peter A. McCue, MD) without clinical information. Staining intensity was scaled as 0 (negative), 1 (weak to moderate positive) and 2 (strong positive).
Patient samples
UM liver metastatic biopsies were formalin fixed and paraffin-embedded immediately following isolation. IHC was performed using anti-phospho-ERBB2 Y1221/Y1222 (6B12) and anti-phospho-cMET Y1234/Y1235 (D26). Staining was scored in a blinded manner, as above. Patient samples were collected under a protocol approved by the IRB at Thomas Jefferson University (IRB protocol number: Control # 11E.548). All patients gave informed consent.
Results
NRG1 and HGF rescue the growth-inhibitory effect of MEK inhibitors in UM cells
We determined the effects of clinically relevant, small molecule MEK inhibitors in three genetically characterized cells lines derived from UM metastases: UM001 was established from liver metastasis and harbors a GNAQ Q209P mutation; UM003 was generated from retroperitoneal metastasis and harbors a GNAQ Q209L mutation (25) and UM004 cells were established from orbital metastasis and harbor a GNAQ Q209P mutation (Supp. Fig. 1A). Based on initial dose response experiments (Supp. Fig. 1B), UM001, UM003 and UM004 cells were treated with 100 nM of the MEK inhibitor, trametinib in subsequent experiments. Trametinib treatment rapidly and persistently blocked ERK1/2 phosphorylation in all cell lines (Fig. 1A). To determine whether MEK inhibition led to growth arrest or a cytotoxic effect, we treated cells with trametinib for 3 days (UM001 and UM004 cells) or 5 days (UM003 cells) and analyzed the cell cycle profile by propidium iodide (PI) staining (Fig. 1B). In all three cell lines, trametinib caused a strong accumulation of the subG1 population (trametinib vs. DMSO: 65% vs. 3% in UM001 cells; 58% vs. 5% in UM003 cells; 23% vs. 0.5% in UM004 cells) and a significant reduction of S-phase population (trametinib vs. DMSO: 1.3% vs. 7% in UM001 cells; 3.5% vs. 1% in UM003 cells; 7.5% vs. 2% in UM004 cells). These results indicate that MEK inhibition elicits cytotoxicity and growth arrest in monocultures of metastatic UM cells.
Figure 1. NRG1 and HGF rescue growth abrogation induced by MEK inhibitors in UM cells.

(A) UM001, UM003 and UM004 cells were treated with 100 nM of trametinib (GSK1120212) for the indicated times. Cell lysates were probed with phospho ERK1/2, total ERK2 and actin antibodies. (B) UM001, UM003 and UM004 cells were treated with DMSO or trametinib for 3 days (UM001 and UM004 cells) or 5 days (UM003). Cells were then fixed, permeabilized and subjected to propidium iodide (PI) staining. Cell cycle analysis was performed with FlowJ software. *P<0.05, **P<0.01, ***P<0.001, based on two-tail Student's t-test assuming unequal variance. (C) UM001 cells were treated with vehicle control, 10 ng/ml of EGF, PDGF-B, HGF, NRG1 and IGF1 alone or together with 100 nM trametinib. After 72 hr, cells were subjected to crystal violet staining. Representative microscopic images of the cells at 200× magnification are shown. Scale bar is equal to 50 μm. (D) UM003 cells were treated as in C for a total of 5 days. Drugs and growth factors were replenished on day 3. Cells were stained with crystal violet.
Since drug resistance to targeted therapies may be mediated by the tumor microenvironment, we sought to identify growth factors that are able to protect metastatic UM cells from MEK inhibition. We screened five growth factors: epidermal growth factor (EGF); platelet-derived growth factor (PDGF); hepatocyte growth factor (HGF); neuregulin 1 (NRG1) and insulin-like growth factor 1 (IGF1) for their capacity to rescue UM cells from MEK inhibition. These growth factors did not enhance proliferation and viability of UM001, UM003 and UM004 cells compared to vehicle-treated cells (Fig. 1C and 1D left panels; Supp. Fig. 2A left panel; Supp. Fig 2B). Treatment of UM cells with trametinib dramatically decreased proliferation and viability, an effect that was blocked by NRG1 and HGF but not EGF and PDGF (Fig. 1C and 1D right panels; Supp. Fig. 2A right panel; Supp. Fig. 2B). IGF1 enhanced proliferation and viability of trametinib-treated UM003 cells, albeit to a lesser extent than NRG1 and HGF (Fig. 1D, right panel; Supp. Fig. 2B, right panel). Next, we tested a second MEK inhibitor, selumetinib/AZD6244, on UM cells. Selumetinib effectively blocked ERK1/2 phosphorylation in these cells (Supp. Fig. 3A). NRG1 and HGF also rescued UM cell survival from selumetinib treatment (Supp. Fig. 3B and Fig. 3C). These data demonstrate that NRG1 and HGF partially restore growth and viability of metastatic UM cells treated with MEK inhibitors.
MEK inhibition enhances responsiveness to NRG1 through ERBB3 and ERBB2 in UM cells
Based on the up-regulation of NRG1-ERBB3 signaling in BRAF V600E cutaneous melanoma cells following RAF inhibitor treatment (27), we tested the impact of trametinib on NRG1-induced ERBB3 signaling in UM cells. Treatment with trametinib sensitized UM001 and UM003 cells to NRG1-stimulated ERBB3 phosphorylation at Y1197 and Y1289 in dose and time course experiments (Fig. 2A–C). Interestingly, UM003, but not UM001 and UM004 cells, showed enhanced expression of ERBB3 in response to trametinib (Supp. Fig. 4A). Phosphorylated Y1197 and Y1298 in ERBB3 are within YXXM-motifs, which dock phosphoinositide 3-kinase (PI3K) leading to AKT phosphorylation (28). In line with enhanced phospho-ERBB3 levels, AKT phosphorylation at S473 and T308 was elevated following NRG1 stimulation in trametinib-treated UM001 and UM003 cells (Fig. 2A–2C). ERBB3 exhibits low intrinsic kinase activity and utilizes a co-receptor, typically another ERBB family member, to signal. UM001, UM003 and UM004 cells express ERBB2 but undetectable levels of EGFR and ERBB4 in either basal or trametinib-treated conditions (Supp. Fig. 4B). NRG1 stimulated ERBB2 phosphorylation in trametinib-treated UM001 and UM003 cells (Fig. 2A–2C). Furthermore, silencing ERBB2 effectively inhibited NRG1-stimulated ERBB3 and AKT phosphorylation in UM001 (Fig. 2D). These data indicate that NRG1-ERBB3/ERBB2 signaling to AKT is elevated in MEK-inhibited, metastatic UM cells.
Figure 2. Trametinib treatment enhances NRG1-ERBB3 signaling in UM cells.

(A) NRG1-ERBB3 signaling is enhanced in cells treated with trametinib. Exponentially growing UM001 cells were treated with vehicle or 100 nM trametinib overnight, followed by treatment with increasing doses of NRG1 for 1 hr. Cells were lysed and phosphorylation of AKT, ERBB3, ERBB2 and ERK1/2 assessed by Western blotting. (B) UM001 cells were treated with vehicle or 100 nM trametinib overnight, followed by treatment 2.5 ng/ml of NRG1 for the indicated time points. Lysates were analyzed as in A. (C) UM003 cells were treated and analyzed as in A. (D) UM001 cells were transfected with 20 nM control siRNA or ERBB2 siRNA. After 72 hr, cells were treated with 100 nM of trametinib overnight, followed by stimulation with 10 ng/ml NRG1 for 1 hr. Knockdown efficiency and phosphorylation of AKT, ERBB3 and ERK1/2 was evaluated by Western blotting.
Targeting NRG1 signaling overcomes resistance to MEK inhibitors in metastatic UM cells
To determine whether blocking ERBB3 prevents NRG1-mediated resistance to trametinib, we took two distinct strategies. First, we utilized U3-1287, a humanized ERBB3 monoclonal antibody that is being used in clinical setting (29). U3-1287 effectively blocked NRG1-stimulated phosphorylation of ERBB3 and downstream AKT activation in UM001 cells (Fig. 3A). Next, we examined the effect of U3-1287 on NRG1-induced resistance to trametinib. As above, cell proliferation (Fig. 3B) and viability (Fig. 3C) of trametinib-treated UM001 cells were partially restored in the presence of NRG1. U3-1287 alone did not suppress the cell growth and viability of UM001 cells but effectively abrogated the protective effect of NRG1 (Fig. 3B & 3C). Based on our evidence that ERBB2 is the co-receptor for NRG1 in UM cells, we utilized lapatinib, a small molecule inhibitor of ERBB2/EGFR. Lapatinib alone did not affect survival of UM001 cells but did dramatically impair the ability of NRG1 to restore growth and viability in trametinib-treated UM001 (Fig. 3D & 3E) and UM003 cells (Fig. 3F). Together, these data demonstrate that targeting NRG1 signaling with ERBB3 antibodies and EGFR/ERBB2 inhibitors overcomes NRG1-mediated resistance to trametinib in metastatic UM cells.
Figure 3. Targeting NRG1 signaling overcomes resistance to trametinib in UM cells.

(A) ERBB3 antibody, U3-1287, blocks NRG1-induced activation of ERBB3 and AKT. UM001 cells were treated with 1 μg/ml or 10 μg/ml of U3-1287 for either 1 hr or 24 hr followed by 10 ng/ml NRG1 stimulation for 15 min. Levels of ERBB3, AKT and ERK1/2 phosphorylation were evaluated by Western blotting with the indicated antibodies. (B) NRG1-induced resistance to trametinib is reversed by U3-1287. UM001 cells were first treated with 100 nM trametinib for 24 hr. Cells were then washed and treated with 10 μg/ml U3-1287 for 45 min, followed by 10 ng/ml NRG1 and 100 nM trametinib for 72 hr. Cells were stained with crystal violet. Representative microscopic images are shown (200 × magnifications). Scale bar is equal to 50 μm (left). (C) UM001 cells were treated as in B. Cell viability was assessed by AlamarBlue® staining. *P<0.05, **P<0.01, ***P<0.001 based on two-tail Student's t-test assuming unequal variance. (D) NRG1-induced resistance was overcome by lapatinib. UM001 cells were treated with 100 nM trametinib, 10 ng/ml of NRG1 or together with 1 μM lapatinib, as indicated for 72 hr. Cells were stained and representative images shown. (E) UM001 cells were treated as in D. Cell viability was assessed by AlamarBlue® staining. *P<0.05, **P<0.01, ***P<0.001, based on two-tail Student's t-test assuming unequal variance. (F) UM003 cells were treated with 100 nM trametinib, 10 ng/ml NRG1 or together with 1 μM lapatinib, as indicated for a total of 5 days. Culture medium was changed and new drug and growth factors were added on day 3. Cell viability was assessed by AlamarBlue® staining. *P<0.05, **P<0.01, ***P<0.001, based on two-tail Student's t-test assuming unequal variance.
HGF induces sustained activation of AKT in trametinib-treated UM cells
Our data show that HGF also effectively protects against MEK inhibitor effects on UM cell growth. To investigate these effects further, UM001 cells were pretreated with trametinib overnight following by stimulation with increasing doses of HGF. In contrast to the effect of NRG1, HGF promoted the initial phosphorylation of cMET and downstream activation of AKT equivalently in trametinib-treated versus vehicle-treated UM001 and UM003 cells (Fig. 4A & 4B). However, HGF-induced phosphorylation of cMET and AKT was maintained at a higher level in trametinib-treated cells at later time points (Fig. 4C). These effects were associated with upregulated cell surface expression of cMET following MEK inhibition in both cell lines (Supp. Fig. 5A). In contrast, IGF1 induced a transient activation of IGF-1R and AKT, irrespective of trametinib treatment, with phosphorylation returning to basal levels at later (>8 hours) time points (Supp. Fig. 5B). Our data suggest that the sustained activation of AKT by HGF may compensate for the loss of ERK1/2 activation in trametinib-treated cells and contributes to resistance to trametinib in UM cells.
Figure 4. HGF signaling induces a sustained activation of AKT and cMET in UM cells.
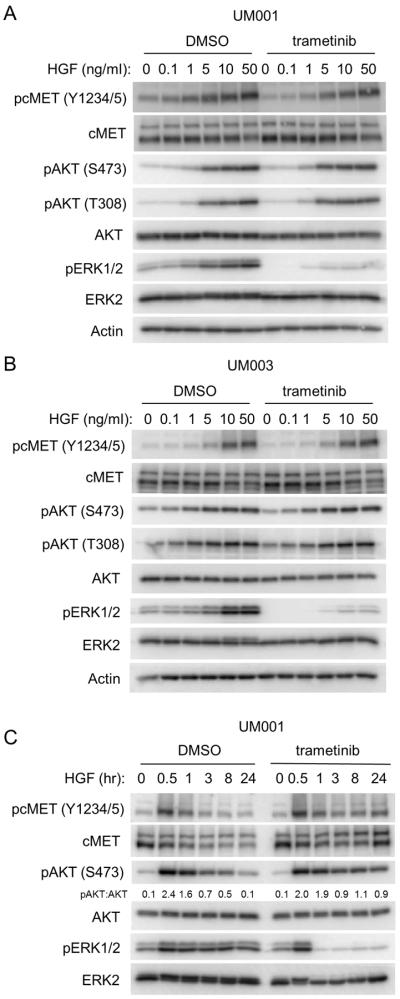
(A) UM001 cells were treated with DMSO or 100 nM trametinib overnight, followed by treatment with increasing doses of HGF for 60 min. Cells were lysed and Western blotted with the indicated antibodies for phosphorylation of AKT, cMET and ERK1/2. (B) UM003 cells were treated and analyzed as in A. (C) HGF stimulation prolongs AKT activation in UM cells. UM001 cells were treated with 10 ng/ml of HGF, or HGF together with 100 nM of trametinib for indicated times. Cells lysates were probed with the indicated antibodies to evaluate phosphorylation of AKT, cMET and ERK1/2.
cMET inhibition overcomes HGF-mediated resistance to trametinib in UM cells
To test whether targeting cMET could abrogate HGF-mediated resistance to trametinib in UM cells, we first used crizotinib, a cMET/anaplastic lymphoma kinase (ALK) inhibitor (30, 31). In UM001 and UM003 cells, crizotinib blocked HGF-induced cMET phosphorylation in a dose dependent manner (Fig. 5A). UM cells were treated with trametinib alone, in combination with HGF and/or crizotinib. At the two doses tested, crizotinib alone did not affect viability (Fig. 5B) or proliferation (Supp. Fig. 6A) of UM001 and UM003 cells. As above, HGF partially restored the viability and cell proliferation of trametinib-treated cells; an effect that was reversed by crizotinib (Fig. 5B & 5C). As a second approach, we tested the effect of silencing cMET expression on sensitivity to trametinib in UM cells. cMET knockdown alone neither altered ERBB3 and ERBB2 levels nor impacted cell growth in UM001 and UM004 cells (Supp. Fig. 6B). Whereas control cells were protected from trametinib-induced inhibition of colony growth by HGF, cMET knockdown cells were sensitive to trametinib despite the presence of HGF (Supp. Fig. 6C). Thus, cMET is required for HGF-mediated resistance to trametinib.
Figure 5. Targeting cMET reverses HGF-induced resistance to trametinib in UM cells.

(A) UM001 and UM003 cells were treated with crizotinib for 4 hr, followed by 10 ng/ml of HGF stimulation for 15 min. Phosphorylation of cMET was evaluated by Western blotting of cell lysates with p-cMET antibody. Actin was used for loading. (B) Exponentially growing UM001 and UM003 cells were treated with DMSO or 100 nM trametinib, in combination with 10 ng/ml HGF and/or crizotinib for 3 days (UM001) and 5 days (UM003). Cell viability was determined by AlamarBlue® staining. **P<0.01 based on two-tail Student's t-test assuming unequal variance. (C) Exponentially growing UM001 and UM003 cells were treated 100 nM trametinib, in combination with HGF and/or crizotinib for 3 days (UM001) and 5 days (UM003). Culture medium was changed and new drug and growth factors were added on day 3). Cells were washed and stained with crystal violet. Images were taken (×200 magnification). (D) UM001 cells were treated with DMSO or 100 nM of trametinib, in combination with 10 ng/ml NRG1 (left) or 10 ng/ml HGF (right) for a total of 3 days. In some conditions, 2 ug/ml MK2206 was also added. Cell growth was determined by crystal violet staining. Images were taken at ×200 magnification. (E) UM001 cells were pretreated with vehicle, 100nM trametinib or 2 μM MK2206 overnight. Cells were then stimulated with 10 ng/ml NRG1 (left) or HGF (right) for 1 hr, as indicated. Activation of AKT, ERK1/2, and TSC2 were analyzed by Western blotting.
AKT inhibition reverses NRG1- and HGF-mediated resistance to trametinib in UM cells
Both NRG1 and HGF promote AKT signaling in trametinib-treated cells. To test whether NRG1 and HGF driven resistance to MEK inhibitors is mediated by AKT, we utilized the inhibitor, MK2206. Addition of MK2206 completely abrogated the protective effect of NRG1 and HGF in trametinib-treated UM001 (Fig. 5D) and UM003 cells (Supp. Fig 7). Notably, MK2206 alone slightly inhibited growth of UM001 cells, while it did not affect the growth of UM003 cells. MK2206 effectively blocked NRG1 and HGF-initiated signaling leading to AKT phosphorylation and downstream AKT targets in trametinib-treated UM001 cells (Fig. 5E). Thus, AKT contributes at least in part to the NRG1 and HGF-mediated protection from MEK inhibitors.
Fibroblast-derived growth factors elicit UM cell resistance to trametinib
Stromal fibroblasts in the tumor microenvironment may promote tumor growth and regulate drug response in a variety of cancer types (32, 33). To determine whether trametinib resistance may be mediated through a paracrine effect from fibroblasts, we examined the activation of ERBB3 and cMET in UM cells by conditioned medium from two fibroblast cell lines: hTERT immortalized BJ1 (HT-BJ1) and Wi38, which produce NRG1 and HGF, respectively (34). Vehicle or trametinib-treated UM001 and UM003 cells were cultured in conditioned medium for 1 hour. Conditioned medium from HT-BJ1 cells induced phosphorylation of ERBB3 and AKT; whereas conditioned medium from Wi38 cells induced phosphorylation of cMET, AKT and ERK1/2 (Fig. 6A). UM001 cells cultured with HT-BJ1 conditioned medium were partially protected from MEK inhibition as assessed by growth (Fig. 6B, left) and viability (Fig. 6B, right). Addition of lapatinib restored the sensitivity to trametinib in these cells (Fig. 6B), consistent with the involvement of ERBB3-ERBB2 signaling. Additionally, we tested whether Wi38 cells promote resistance to trametinib in UM cells through HGF-cMET signaling. UM001 cells growing in Wi38 conditioned medium were resistance to trametinib, an effect reversed by addition of crizotinib (Fig. 6C). These data support the notion that paracrine effects of NRG1 and HGF from fibroblasts promote resistance to trametinib, which is overcome with agents specifically targeting ERBB3/ERBB2 and cMET pathways in human metastatic UM cells.
Figure 6. Paracrine effects of NRG1 and HGF from fibroblasts drives resistance to trametinib in UM cells.

(A) UM001 and UM003 cells were cultured for 1 hr in unconditioned growth medium or fibroblast conditioned medium (CM) collected from either HT-BJ1 cells or Wi38 cells. Cells treated with 10 ng/ml of NRG1 and HGF were used as control. Activation of ERBB3, cMET, AKT and ERK1/2 were analyzed by Western blotting. (B) UM001 cells were cultured in conditioned medium collected from HT-BJ1 cells (CM) or unconditioned medium (non-CM). Cells were treated with 100 nM trametinib ± 1 μM lapatinib, as indicated. After 72 hr, cells were stained with crystal violet. Representative microscopic images were shown with a 200× magnification. Scale bar is equal to 50 μm (left). Cell viability was also assessed by AlamarBlue® staining after 72 hr (right). (C) UM001 cells were cultured in conditioned medium collected (CM) from Wi38 cells or unconditioned medium (non-CM), as a control. Cells were treated with 100 nM trametinib and 0.5 μM crizotinib as indicated. After 72 hr, cells were stained with crystal violet. Representative microscopic images were shown with a 200× magnification. Scale bar is equal to 50 μm (left). AlamarBlue® staining was also performed to determine viability (right).
ERBB2 and cMET are activated in orthotopic UM xenografts and UM hepatic metastases
To determine whether ERBB3/ERBB2 complexes and cMET are activated in vivo, we first examined UM xenografts. UM001 cells were injected directly into the liver of NOD SCID gamma (NSG, NOD.Cg-PrkdcSCIDIl2rgtm1Wjl/SzJ) mice and were allowed to grow for 4–5 weeks. Inoculated UM001 cells grew and developed intrahepatic metastases with a high (18/19) success rate. UM001 xenografts growing in NSG mice stained positive for phospho ERBB2 (Fig. 7A, left). Due to the incompatibility of HGF across species (35), we implanted UM001 cells into hHGFki mice, in which the cDNA of human HGF was knocked into the mouse HGF locus by homologous recombination for analysis of phospho cMET. UM001 xenografts growing in hHGFki mice stained positive for phospho cMET (Fig. 7A, right).
Figure 7. Activation of cMET and ERBB2 in UM xenografts and liver metastases of UM patients.

(A) UM001 cells (1 × 106) were injected into the liver of NSG mice or hHGF-ki mice and allowed for growth for 4–5 weeks (NSG mice) or 8 weeks (hHGFki mice). Tumor tissues were fixed embedded and sections were stained with IgG isotype control, anti-phospho ERBB2 and anti-phospho cMET. Representative images are shown at x400 magnification. (B) Biopsies from liver metastases from seven UM patients were stained for anti phospho-ERBB2 and anti phospho-cMET. Staining intensity was scored 0 (no staining), 1 (weak to modest staining) and 2 (strong staining). The percentage of tumor cells was semi-quantitated. (C) Representative images (x400 magnification) of phospho-ERBB2 (top panel) and phospho-cMET (bottom panel) staining with differing intensities in liver metastases of UM patients are shown.
We next extended our study to analyze liver metastases of UM patients (Supp. Table 1). Biopsies from seven UM patients with liver metastasis were stained with anti-phospho ERBB2 and anti-phospho cMET antibodies. Staining intensity was scored 0 (negative staining), 1 (weak to medium positive staining) and 2 (strong positive). Percentage of tumor cells was semi-quantitated. We observed positive staining of phospho ERBB2 in all seven samples and positive staining of phospho cMET in five out of seven samples (Fig. 7B). Representative images with various staining intensity were shown in Fig. 7C. These data suggest that cMET and ERBB2 are activated in UM in the liver metastatic microenvironment.
Discussion
The response of genetically-defined tumors to a targeted therapy is typically heterogeneous. Many patients display no tumor shrinkage and are regarded as exhibiting primary/intrinsic resistance mediated by either pre-existing (innate) or rapid adaptive response mechanisms. Similar mechanisms may be present, albeit to a lesser extent, in tumors that effectively respond to targeted therapies and are likely to modulate the timing of acquired resistance. Here, we describe that both innate and adaptive mechanisms occur in mutant GNAQ metastatic UM cells responding to clinical-grade MEK inhibitors.
Our studies utilize GNAQ mutant human metastatic UM cell lines. These represent an important resource to the field given the high percentage of UMs harboring GNAQ or GNA11 mutations, the noted lack of available cell lines for UM (36), the concern that lines may be cutaneous melanoma (37) and the common use of lines derived from non-metastatic lesions for drug response studies in the UM literature. Using mutant GNAQ metastatic UM lines, we show that clinical grade MEK inhibitors (trametinib and selumetinib) block cell growth in vitro by either promoting cell death or inducing a proliferative arrest. This heterogeneous response is consistent with data from the Woodman group who showed that trametinib promoted >10% increase in apoptosis in two out of six GNAQ/GNA11 uveal melanoma cell lines (19). We identify two growth factors, NRG1 and HGF, which are able to reverse the cytotoxic and growth inhibitory effects of trametinib and selumetinib. However, the actions of NRG1 and HGF on their cognate receptors, ERBB3 and cMET, differ. NRG1 activation of ERBB3 is enhanced in MEK-inhibited cells in an adaptive manner. By contrast, initial HGF activation of cMET is comparable in untreated and MEK inhibitor-treated cells, although is more persistent in the MEK-inhibited cells. These data are similar to findings in cutaneous mutant BRAF melanoma cells, in which NRG1 adaptively up-regulate ERBB3-AKT signaling in response to vemurafenib/PLX4720 (27) and in which HGF promotes resistance via adaptive and innate mechanisms depending on the cell line (34).
Both ERBB3 and cMET activate the PI3K-AKT pathway and the addition of a PI3K inhibitor to MEK treatment enhances apoptosis in mutant GNAQ cells in vitro (19). Furthermore, our data show that targeting AKT reverses growth factor-mediated resistance. It has long been recognized that the combination of ERK1/2 and PI3K pathway inhibitors will likely be beneficial in many tumor settings; however, the advancement of PI3K and AKT targeting agents is currently limited in the clinic by toxicity issues and poor target inhibition. An additional concern is that ERBB3 expression/activity is frequently up-regulated as a compensatory feedback mechanism to PI3K inhibitors (38, 39).
There is growing appreciation for the need for combinatorial targeted therapy studies. For example, recent preclinical studies show that combined inhibition of MEK and PKC improves efficacy compared to treatment with either single agent in GNAQ/11 mutant UM (40, 41). We tested the effect of co-targeting the receptors, ERBB3 or cMET, in combination with MEK inhibitors. Targeting the ERBB3/ERBB2 complex with U3-1287/AMG88 or lapatinib effectively reversed the NRG1-mediated resistance to MEK inhibitors. U3-1287 is one example of a humanized ERBB3 antibody that has entered early phase clinical trials (29, 42). Similarly, we targeted cMET either with crizotinib or by RNA interference. Crizotinib is an ATP-competitive inhibitor of cMET as well as ALK and is FDA-approved for non-small cell lung carcinoma patients harboring ALK gene fusions. Targeting cMET reversed HGF-mediated protection from MEK-inhibitor induced growth blockade. Others have utilized the cMET inhibitor, MK-8033, to inhibit growth in mutant GNAQ UM cells (43). Overall, these data highlight that NRG1 and/or HGF-mediated resistance may underlie the modest response rate to MEK inhibitors in metastatic UM. Furthermore, our findings suggest that targeting ERBB3 and/or cMET may enhance the effect of MEK inhibitor in advanced-stage, mutant GNAQ UM patients.
Low levels of phosphorylated ERBB3 and cMET were detected in the absence of NRG1 and HGF, respectively, indicating that these ligands are poorly expressed by tumor cells. UM frequently metastasizes to the liver, a tissue in which both NRG1 and HGF are readily detected (44, 45), highlighting the possibility that these growth factors mediate resistance to MEK inhibitors via paracrine action. To this end, we tested the effect of stromal-produced growth factors on UM cell resistance to MEK inhibitors. Wi38 and BJ1 fibroblasts produce high levels of NRG1 and HGF, respectively, and conditioned medium from these cells promoted AKT phosphorylation and growth in MEK-inhibited UM cells in a manner dependent on the cognate receptor. These findings are similar to the notion that fibroblast-derived HGF protects against RAF inhibitors in cutaneous melanoma (34) and add to growing evidence for factors in the tumor microenvironment being able to modulate the response to targeted anticancer agents. Furthermore, our in vivo data from both a UM cell liver colonization model and liver metastatic patient samples show that the ERBB3/ERBB2 and cMET receptors are frequently phosphorylated.
In summary, we have identified that the growth factors, NRG1 and HGF, mediate resistance to MEK inhibitors in metastatic UM cells. Targeting NRG1 or HGF signaling overcomes the resistance elicited by these growth factors. We have also provided evidence that paracrine effects of NRG1 and HGF from fibroblasts protect UM cells from MEK inhibition. These data provide new insights into the mechanisms that regulate resistance to MEK inhibitors in metastatic UM. On-going efforts are focused on utilizing clinical grade anti-ERBB3 and anti-cMET monoclonal antibodies in combination with MEK inhibitors in pre-clinical studies.
Supplementary Material
Acknowledgements
U3-1287/AMG888 was generously provided by U3-Pharma GmbH (Martinsried, Germany). We thank Dr. Ubaldo Martinez-Outschoorn for the immortalized foreskin fibroblastic BJ1 cell line. This project was funded by a Dean's Transformative Science Award, a TJU Programmatic Initiative Award and NIH R01 CA160495. The Sidney Kimmel Cancer Center core facilities are supported by National Institutes of Health/National Cancer Institute Support Grant (2 P30 CA056036-13).
Footnotes
Conflict of interest: No potential conflicts of interest were disclosed by the authors.
References
- 1.Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118:1881–5. doi: 10.1016/j.ophtha.2011.01.040. [DOI] [PubMed] [Google Scholar]
- 2.Luke JJ, Triozzi PL, McKenna KC, Van Meir EG, Gershenwald JE, Bastian BC, et al. Biology of advanced uveal melanoma and next steps for clinical therapeutics. Pigment Cell Melanoma Res. 2014;28:135–47. doi: 10.1111/pcmr.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cruz F, 3rd, Rubin BP, Wilson D, Town A, Schroeder A, Haley A, et al. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 2003;63:5761–6. [PubMed] [Google Scholar]
- 4.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Cohen Y, Goldenberg-Cohen N, Parrella P, Chowers I, Merbs SL, Pe'er J, et al. Lack of BRAF mutation in primary uveal melanoma. Invest Ophthalmol Vis Sci. 2003;44:2876–8. doi: 10.1167/iovs.02-1329. [DOI] [PubMed] [Google Scholar]
- 6.Weber A, Hengge UR, Urbanik D, Markwart A, Mirmohammadsaegh A, Reichel MB, et al. Absence of mutations of the BRAF gene and constitutive activation of extracellular-regulated kinase in malignant melanomas of the uvea. Lab Invest. 2003;83:1771–6. doi: 10.1097/01.lab.0000101732.89463.29. [DOI] [PubMed] [Google Scholar]
- 7.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–9. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harbour JW. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res. 2012;25:171–81. doi: 10.1111/j.1755-148X.2012.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onken MD, Worley LA, Long MD, Duan S, Council ML, Bowcock AM, et al. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49:5230–4. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shoushtari AN, Carvajal RD. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014;24:525–34. doi: 10.1097/CMR.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 12.Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933–6. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3:1122–9. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45:133–5. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–3. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewens KG, Kanetsky PA, Richards-Yutz J, Purrazzella J, Shields CL, Ganguly T, et al. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Invest Ophthalmol Vis Sci. 2014;55:5160–7. doi: 10.1167/iovs.14-14550. [DOI] [PubMed] [Google Scholar]
- 17.Dono M, Angelini G, Cecconi M, Amaro A, Esposito AI, Mirisola V, et al. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br J Cancer. 2014;110:1058–65. doi: 10.1038/bjc.2013.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaque JP, Dorsam RT, Feng X, Iglesias-Bartolome R, Forsthoefel DJ, Chen Q, et al. A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol Cell. 2013;49:94–108. doi: 10.1016/j.molcel.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, et al. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012;18:4345–55. doi: 10.1158/1078-0432.CCR-11-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambrosini G, Musi E, Ho AL, de Stanchina E, Schwartz GK. Inhibition of mutant GNAQ signaling in uveal melanoma induces AMPK-dependent autophagic cell death. Mol Cancer Ther. 2013;12:768–76. doi: 10.1158/1535-7163.MCT-12-1020. [DOI] [PubMed] [Google Scholar]
- 21.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–9. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311:2397–405. doi: 10.1001/jama.2014.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 24.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida M, Selvan S, McCue PA, DeAngelis T, Baserga R, Fujii A, et al. Expression of insulin-like growth factor-1 receptor in metastatic uveal melanoma and implications for potential autocrine and paracrine tumor cell growth. Pigment Cell Melanoma Res. 2014;27:297–308. doi: 10.1111/pcmr.12206. [DOI] [PubMed] [Google Scholar]
- 26.Boisvert-Adamo K, Aplin AE. B-RAF and PI-3 kinase signaling protect melanoma cells from anoikis. Oncogene. 2006;25:4848–56. doi: 10.1038/sj.onc.1209493. [DOI] [PubMed] [Google Scholar]
- 27.Abel EV, Basile KJ, Kugel CH, 3rd, Witkiewicz AK, Le K, Amaravadi RK, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. 2013;123:2155–68. doi: 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res. 2010;16:1373–83. doi: 10.1158/1078-0432.CCR-09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LoRusso P, Janne PA, Oliveira M, Rizvi N, Malburg L, Keedy V, et al. Phase I study of U3-1287, a fully human anti-HER3 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2013;19:3078–87. doi: 10.1158/1078-0432.CCR-12-3051. [DOI] [PubMed] [Google Scholar]
- 30.Zillhardt M, Christensen JG, Lengyel E. An orally available small-molecule inhibitor of c-Met, PF-2341066, reduces tumor burden and metastasis in a preclinical model of ovarian cancer metastasis. Neoplasia. 2010;12:1–10. doi: 10.1593/neo.09948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–17. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 32.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–50. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 33.Ostman A. The tumor microenvironment controls drug sensitivity. Nat Med. 2012;18:1332–4. doi: 10.1038/nm.2938. [DOI] [PubMed] [Google Scholar]
- 34.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhargava M, Joseph A, Knesel J, Halaban R, Li Y, Pang S, et al. Scatter factor and hepatocyte growth factor: activities, properties, and mechanism. Cell Growth Differ. 1992;3:11–20. [PubMed] [Google Scholar]
- 36.Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, et al. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012;25:182–7. doi: 10.1111/j.1755-148X.2012.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu X, Ambrosini G, Roszik J, Eterovic AK, Stempke-Hale K, Seftor EA, et al. Genetic Analysis of the 'Uveal Melanoma' C918 Cell Line Reveals Atypical BRAF and Common KRAS Mutations and Single Tandem Repeat Profile Identical to the Cutaneous Melanoma C8161 Cell Line. Pigment Cell Melanoma Res. 2015;28:357–9. doi: 10.1111/pcmr.12345. [DOI] [PubMed] [Google Scholar]
- 38.Chakrabarty A, Sanchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A. 2012;109:2718–23. doi: 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2013;33:4724–34. doi: 10.1038/onc.2013.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sagoo MS, Harbour JW, Stebbing J, Bowcock AM. Combined PKC and MEK inhibition for treating metastatic uveal melanoma. Oncogene. 2014;33:4722–3. doi: 10.1038/onc.2013.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aurisicchio L, Marra E, Roscilli G, Mancini R, Ciliberto G. The promise of anti-ErbB3 monoclonals as new cancer therapeutics. Oncotarget. 2012;3:744–58. doi: 10.18632/oncotarget.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chattopadhyay C, Grimm EA, Woodman SE. Simultaneous inhibition of the HGF/MET and Erk1/2 pathways affect uveal melanoma cell growth and migration. PLoS One. 2014;9:e83957. doi: 10.1371/journal.pone.0083957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh SY, He JR, Hsu CY, Chen WJ, Bera R, Lin KY, et al. Neuregulin/erythroblastic leukemia viral oncogene homolog 3 autocrine loop contributes to invasion and early recurrence of human hepatoma. Hepatology. 2011;53:504–16. doi: 10.1002/hep.24083. [DOI] [PubMed] [Google Scholar]
- 45.Maher JJ. Cell-specific expression of hepatocyte growth factor in liver. Upregulation in sinusoidal endothelial cells after carbon tetrachloride. J Clin Invest. 1993;91:2244–52. doi: 10.1172/JCI116451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.