

Abstract
The discovery that two common APOL1 alleles were strongly associated with non-diabetic kidney diseases in African descent populations led to hope for improved diagnosis and treatment. Unfortunately, we still do not have a clear understanding of the biological function played by APOL1 in podocytes or other kidney cells, nor how the renal risk alleles initiate the development of nephropathies. Important clues for APOL1 function may be gleaned from the natural defense mechanism of APOL1 against trypanosome infections and from similar proteins (e.g. diphtheria toxin, mammalian Bcl-2 family members). This review provides an update on the biological functions for circulating (trypanosome resistance) and intracellular (emerging role for autophagy) APOL1. Further, we introduce a multimer model for APOL1 in kidney cells that reconciles the gain-of-function variants with the recessive inheritance pattern of APOL1 renal risk alleles.
Keywords: APOL1, renal function, innate defense, autophagy, multimer, toxicity
The discovery in 2010 that the common APOL1 G1 and G2 genetic variants are strongly associated with glomerular disease in African descent populations has opened a door that may lead to improved understanding and treatments.1 Unfortunately, the door has been more difficult for researchers to walk through than many expected. Despite the extensive efforts deployed by the nephrology community, we still do not have a good understanding of how these variants injure podocytes or other kidney cells. This review provides an update on the biological functions for the circulating and intracellular APOL1 forms. We also propose a model for APOL1 renal function (APOL1 multimer weapon with a trigger-lock mechanism for safety) that reconciles the gain of apparent deleterious function with the recessive inheritance pattern of APOL1 risk alleles.
The APOL family
APOL1 is one of the 6 members from the APOL gene family (APOL1-6), organized in a cluster on human chromosome 22.2–4 APOL gene homologues are found through out the animal kingdom. The APOL gene family arose by gene duplication in primates, but only humans, gorillas and baboons retained a functional, expressed APOL1 gene. The function of the apolipoproteins L (APOLs) is largely unknown. APOL1 was initially discovered complexed in high-density lipoprotein 3 (HDL-3) particles, which were identified as the key component of trypanolytic factor (TLF) in human serum.5 The exploration of APOL1 trypanolytic activity revealed an organization in three domains: a pore-forming domain, a pH-sensitive membrane-addressing domain and an SRA-interacting domain (Figure 1). The organization of the pore-forming domain directly adjacent to a membrane-addressing domain is similar to that of bacterial colicins, diphtheria toxin and mammalian Bcl-2 family members.6,7 All APOLs are closely related, although the APOL5 and APOL6 genes are evolutionary divergent from the APOL1-4 gene cluster;3 it is predicted that the 3 domain organization is conserved in all the APOL family members.
Figure 1. Predicted APOL1 structural domain organization.

Pore-forming domain covers residues 60 to 235; BH3 domain = 154–168; membrane-addressing domain = 238–304; and SRA-interacting domain = 339–398. BH3, Bcl-2 homology domain 3.
APOL1 is the only secreted member of the family, having acquired an N-terminal signal peptide. The circulation of APOL1 in HDL-3 particles suggests a role in lipid transport and metabolism,3,8,9 which could be critical in maintaining the plasma membrane of the extensive foot processes of podocytes. However, it remains unclear if APOL1-mediated renal injury is initiated by endogenous kidney-expressed APOL1 or by circulating APOL1. A recent report proposed that the high level of APOL1 protein expression in normal human podocytes is due to both endogenous synthesis and uptake from the circulation,10 and uptake of APOL1 G1 and G2 renal risk isoforms was shown to contribute to human podocyte injury.11 These in vitro findings contrast with two renal allograft studies that suggested that kidney-expressed APOL1, but not circulating APOL1, damages kidneys: allograft survival was not affected by recipient APOL1 genotype suggesting no impact of recipient circulating APOL1;12 however kidneys from donors with two APOL1 risk alleles had significantly shorter survival time in recipient compared to kidneys from donors carrying one or no risk allele, suggesting a role for donor kidney-endogenous APOL1.13 Further studies are needed for a definitive answer –particularly renal allograft studies where the APOL1 genotype of both the kidney donor and kidney recipient are known.
APOL1 and resistance to trypanosome infection
APOL1 is the trypanolytic toxin providing innate resistance against Trypanosoma brucei infection, which causes animal and African human trypanosomiasis (African sleeping sickness) in many mammalian species, including African primates.7,14 The parasite internalizes the APOL1-containing TLF through both fluid phase and receptor-mediated endocytosis15 and the particle is delivered to the lysosome through the endocytic pathway. The progressive acidification of the environment triggers conformational changes in the membrane-addressing domain of APOL1 resulting in the release of APOL1 from the HDL particle within the lysosomal membrane, where APOL1 forms an ionic channel.6,16 The ion influx then provokes osmotic swelling and death of the parasite. In order to replicate in their hosts, trypanosomes have evolved different mechanisms to lock the trigger of the APOL1 lethal weapon: T.b. rhodesiense evolved a serum resistance associated (SRA) glycoprotein that binds to APOL1 within the lysosome to abrogate its toxicity,7 whereas T.b. gambiense-specific glycoprotein (TgsGP) forms hydrophobic β-sheets that stiffen the endo-lysosomal membrane to prevent APOL1 membrane insertion and toxicity.17 In addition, to fully evade APOL1-mediated trypanolysis, APOL1 uptake is limited and APOL1 degradation is enhanced in T.b. gambiense.
The APOL1 G1 and G2 renal risk alleles are located in the SRA-interacting domain (Figure 1) and restore APOL1-mediated protection against T.b. rhodesiense to prevent acute trypanosomiasis in humans.1,18 APOL1 G1 and G2 variant isoforms are both potent killers of T.b. rhodesiense, but intriguingly, they seem to act via different trypanolytic mechanisms (Table 1). The G1 allele is composed of two SNPs in near-perfect linkage disequilibrium (G1G, p.S342G and G1M, p.I384M) but only G1G can kill trypanosome in vivo.18 Contrary to the G2 deletion, G1G does not occur within the epitope (leucin zipper 370–392) that is essential for optimal SRA-binding and trypanolytic activity.19 As a consequence, G2 has a reduced affinity for trypanosome SRA, but not G1G whose affinity for SRA is similar to the wild-type isoform.1,18 G1G is located in a putative α-helix domain and has been hypothesized to stabilize the membrane association of the protein isoform.18 The G1 and G2 isoforms tend to induce cell death and tissue injury,11,20 but with different levels of toxicity: G2 is trypanolytic at a lower titer than G1 in vitro (104-fold dilution vs. undiluted),1 and G1 induces widespread severe liver necrosis in mice whereas G2 caused focal and moderate necrosis.18 The trypanolytic mechanics appear different for G1 and G2, yet they both efficiently kill T.b. rhodesiense and exhibit equivalent effect size for FSGS/HIVAN when comparing individuals carrying G1/G1, G2/G2, and G1/G2 (Table 1).21
Table 1.
APOL1 G1 and G2 risk alleles for FSGS are efficient T.b. rhodesiense killers through different trypanolytic mechanisms.
| Trypanolytic activity | Toxicity | ||||||
|---|---|---|---|---|---|---|---|
| APOL1 isoform | T.b. brucei | T.b. gambiense | T.b. rhodesiense | Trypanosome SRA binding | Dilution factor for 100% in vitro lysis | Mice liver necrosis | OR [95%CI] for FSGS, recessive model |
| WT (G0) | Lysis | No | No | Yes | – | None | Ref. |
| G1G | Lysis | No | Lysis | Yes | 101 or undiluteda | Severe and widespread | 17 [10–32]b |
| G1M | Lysis | No | No | Yes | None | ||
| G2 | Lysis | No | Lysis | No | 104 | Moderate and Focal | 25 [9–82]c |
APOL1 in immunity
Additional studies suggest that APOL1 might play a broader protective role in innate immunity since (1) APOL genes are upregulated by pro-inflammatory cytokines such as IFNγ and TNF,3,11,20,22–25 (2) APOL1 can ameliorate Leishmania parasitic infection,26 and (3) restrict HIV-1 in vitro replication in macrophages.24
The link between inflammation and APOL1 expression may constitute a modifying factor that might explain the incomplete penetrance of the G1/G2 variants for chronic kidney disease, i.e. why only a fraction of individuals carrying two renal risk alleles will develop nephropathy. In particular, the high penetrance of the risk alleles in HIV collapsing nephropathy may be due to elevated and persistent IFNγ levels in response to the virus.11,27 Interactions of APOL1 with other genes or other non-HIV viral infections might also act as second hits.28
APOL1 and programmed cell death
Among the suggested mechanisms by which APOL1 contributes to glomerulosclerosis are apoptosis,5,29,30 autophagy,25,30–32 or endocytosis and lysosomal stimulation.24 All APOLs contain a putative Bcl-2 homology domain 3 (BH3) within the pore-forming domain (Figure 1). Most BH3-only proteins are activators of programmed cell death,33,34 and in accordance, APOL6 was shown to induce apoptosis,30,35 and APOL1 can initiate autophagic cell death under certain circumstances.25,31 This accumulation of evidence combined with the emergence of autophagy as a major pathway in kidney function and glomerular disease36–40 provides a promising avenue of research for revealing the pathophysiological mechanism of APOL1-mediated renal cell injury. A recent study reported that over-expression of APOL1 G1 and G2 variants in human podocytes drove enhanced lysosomal membrane permeability and cell death.11 It is notable that both the trypanolytic activity of secreted APOL1 and the autophagy function of intracellular APOL1 converge on endosome and lysosome trafficking, which is coherent with the pH-dependent membrane-addressing function of APOL1.
APOL genes evolution, pressure of selection and binding partners
Functional APOL1 was only identified in humans, gorillas, baboons and possibly a few other African primates, but was either completely lost (e.g. chimpanzee) or pseudogenized (e.g. orangutan, macaque) in other African primates, suggesting a fitness cost greater than the benefit conferred against extracellular parasites.14,29,41 In primates, APOLs evolved rapidly and were under positive selection by pathogens (hence reinforcing the likely broader role for these genes in immunity), especially in the functional C-terminal region.29 For this reason, this domain is thought to be fundamental for APOL function and it is predicted that molecules interacting with the C-terminal domain are essential for preventing APOL-mediated cell death, similar to the APOL1/SRA system in trypanosomes.19,29 Vanhollebeke and Pays have speculated that mammalian ‘SRA-like’ proteins are involved in the natural control of APOL toxicity through interaction with the C-terminal helix,5 and Wan et al. have postulated that APOL1 and APOL6 interacting partners, either protein or lipid, might mediate different death-signaling pathways.32 Efforts to identify APOL1 binding partner(s) have been initiated to reveal the regulatory mechanisms of APOL1 toxicity that could explain kidney injury. The action of a second factor regulating APOL1-mediated cell death could explain why only kidney cells seem to be damaged from G1 and G2 risk alleles when APOL1 expression is quite ubiquitous: the second factor could have a different level of expression in non-kidney cells, or a similar factor with a stronger affinity for APOL1 might be expressed in other cell types to lock its deleterious function. Wan et al. demonstrated that APOL1 can bind in vitro with high affinity to lipids involved in cell death regulation: phosphatidic acid and cardiolipin that are associated with mTOR/rapamycin autophagy signaling and mitochondria-mediated apoptosis, respectively.32 Sedor and colleagues recently used a secondary structure-based strategy to identify human proteins with similar structure to trypanosomal SRA and identified VAMP8, YKT6, osteocalcin, VAMP1 and SEC22b as top candidates (O’Toole JF, et al., SA-OR095, ASN annual meeting, Atlanta, GA, 2013; Sedor JR, invited presentation, ASN annual meeting, Philadelphia, PA, 2014; Sedor JR, personal communication). They further explored VAMP8, a SNARE protein of the endo-lysosomal compartment that can anchor SNARE proteins from the autophagosome to trigger fusion between the two compartments.42 By co-immunoprecipitation and surface plasmon resonance experiments, VAMP8 was shown to interact with APOL1 in a variant-dependent manner to regulate APOL1 toxicity: compared to G0 (wild-type), the APOL1-VAMP8 interaction was reduced with G1 and G2 protein isoforms, which impacted APOL1-induced autophagy. Furthermore, molecular dynamics simulations revealed that G1 and G2 are helix-stabilizing variants, and that the G1 and G2 isoforms have less conformational mobility than G0, which would explain why the interaction of APOL1 binding partners would be impaired with G1 and G2 isoforms (Sedor JR, invited presentation, ASN annual meeting, Philadelphia, PA, 2014; Sedor JR, personal communication). Whether phosphatidic acid, cardiolipin and VAMP8 regulate APOL1-induced autophagy or cell death in kidney cells and whether other APOL1 binding partners could modulate any APOL1-related function have not yet been formally demonstrated or related to kidney injury.
APOL1 renal function and the recessive model
Beyond the lack of clarity surrounding the perturbation of kidney cellular function by the G1 and G2 isoforms, any mechanism for renal cell injury must account for the strong recessivity observed for APOL1 renal risk alleles in epidemiological studies (discussed in43). Usually, a recessive model correlates with a loss-of-function mutation. However, APOL1 is not required for kidney development or kidney homeostasis as most mammalian species, including higher primates, lack APOL1. Indeed, an Asian Indian who is a homozygous null for APOL1 was shown to have normal renal function, as did members of his family who were heterozygous carriers of the null mutation.44,45 This and the evolutionary history of APOL1 suggest that APOL1 exerts a redundant function. Therefore, if we consider a gain of deleterious function model for G1 and G2, we would expect that an additive or dominant model would best fit the association with glomerular disease (Figure 2, left panel): carrying one copy of the G1 or G2 variant should drive a damaging phenotype, as detrimental as carrying two copies (dominant), or an intermediate phenotype (additive), which is in contradiction with all evidence pointing to a largely recessive model.43
Figure 2. Model of APOL1 multimers in renal cells reconciling a gain of deleterious function model with the recessive pattern of inheritance.
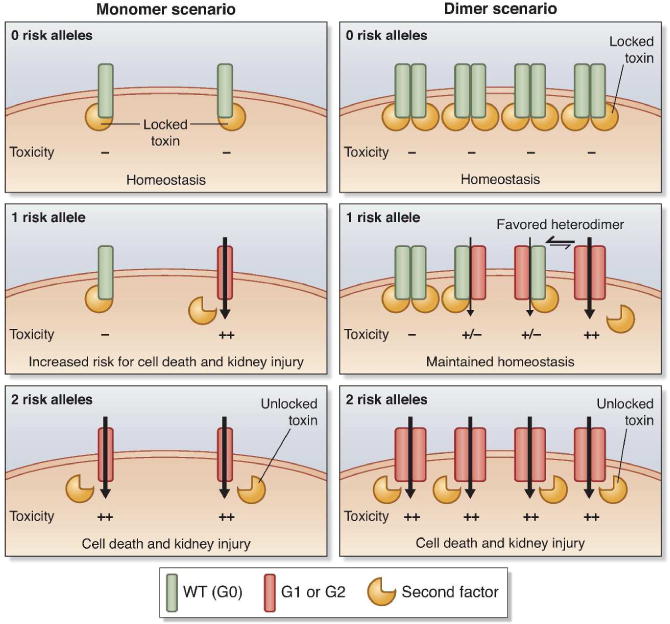
Monomer and dimer scenarii are depicted on the left and right panels, respectively. Similarly to its trypanolytic function, we consider that APOL1 wild-type (G0, in green) toxic activity can be inhibited by a yet to be formally identified second factor (the trigger-lock system, in yellow) protecting the cell from death (locked weapon). The inhibition is lifted (actuating the trigger) by the G1 and G2 isoforms (in dark orange), enhancing the toxicity (illustrated by the black arrow) and cell death (weapon discharge) that could eventually lead to the development of glomerular injury. Only the dimer scenario offers a neutral activity for the carriers of one risk allele by limiting the number of damaging APOL1 channels (heterodimers exhibit a low or no toxicity owing to the second factor binding) and therefore reconciles with the recessive model of inheritance.
Multimerization of APOL1
Based on the multimerization of similar proteins, we propose a model of multimerization for APOL1 in renal cells to reconcile the recessive pattern of inheritance with a gain of deleterious function model (Figure 2). As previously mentioned, APOL domain organization share some structural and functional similarities with bacterial colicins, diphtheria toxin and mammalian Bcl-2 family members (a pore-forming domain adjacent to a pH-sensitive membrane-addressing domain) suggesting a similar activity for all these proteins. Remarkably, the diphtheria toxin pathway through the infected cell is highly comparable to APOL1’s pathway in the trypanosome. The diphtheria toxin enters the cell by endocytosis and then travels through the endosome, where the progressive acidification of the environment triggers a conformational change in the membrane-addressing domain, which allows the insertion of the toxin into the membrane, the formation of a ionic pore, and the translocation of the toxic protein fragment into the cytoplasm.46 Interestingly, diphtheria toxins are able to multimerize in membranes,47–49 and some bacterial colicins also seem capable of dimerization.50–52 Finally, dimerization and multimerization are essential for the mammalian Bcl-2 family members harboring a BH3 domain to exert their pro-apoptotic activity.53–57 In light of this body of evidence in analogous proteins, the multimerization of APOL1 proteins is plausible and warrants investigation. We therefore ran simulations to estimate the recessivity for monomers and multimers from dimers to hexamers (Figure 3). In this model, we considered that APOL1 toxicity is antagonized by a second factor (the lipid or protein trigger-lock system, see above) interacting with the C-terminal domain of APOL1 wild-type isoforms, and that this inhibition is either attenuated or abrogated by the G1 and G2 isoforms, due to loss of binding affinity for G1 and/or G2 with the second factor. For multimers carrying one or more wild-type APOL1 isoforms, binding and blocking of toxicity are retained. Our simulations show that multimerization would fit a completely or almost completely recessive model –as observed for G1/G2 associations with CKD and ESKD–, when a monomer-only model would fit an additive model, in contradiction with the epidemiological data.
Figure 3. Modeling recessivity as a function of multimerization and loss of binding to the trigger-lock.

We assume the following simple model: 1) There is a decrease of probability of having the trigger-lock bound to APOL1 for APOL1 risk isoforms (G1 and G2 are assumed to have equal binding) compared to wild-type (WT); 2) For carriers of 1 risk allele (WT/G1 or WT/G2 heterozygotes), multimers are formed from WT or risk molecules, drawn randomly to give a binary distribution; 3) Binding of the trigger-lock to any of the APOL1 molecules in the multimer blocks multimer activity. For this model, we plot the dominance coefficient as a function of the loss of binding probability for risk allele isoforms to the trigger-lock (expressed as a % of WT binding), from monomers to hexamers. Here, the dominance coefficient ranges from 0.5 predicting an additive model (where one risk allele carriers have half the increased risk of kidney injury of two risk alleles carriers) to 0 for a completely recessive model (where one risk allele carriers have no increased risk).
If we consider that APOL1 exerts its function in a monomeric form (Figure 2, left panel), then we can posit that homozygosity for the wild-type (or G0) allele (0 risk allele) will maintain podocyte (or other renal cell) homeostasis due to the interaction with the second factor that limits toxicity and protects from cell death (locked weapon with the safety engaged). In contrast, individuals with 2 risk alleles would have a decreased affinity for the second factor (actuate the trigger), enhancing the toxicity and cell death that could eventually lead to the development of glomerular injury (weapon discharge). However, if this model were correct, heterozygosity for G1 or G2 (1 risk allele) would also display an increased toxicity that should drive an intermediate phenotype, illustrated by an additive model of inheritance, which is in contradiction with clinical data.
In a dimer scenario (Figure 2, right panel), individuals with no risk allele would maintain cell homeostasis (locked weapon) while individuals carrying two risk alleles would exhibit a high level of podocyte cell death leading to renal injury (weapon discharge). In the case of one risk allele, APOL1 could form homodimers for the wild-type protein, homodimers for the risk protein, and heterodimers for the wild-type and renal risk isoforms. Heterodimer-mediated toxicity would likely be reduced or abrogated by the interaction with the second factor. Individuals with one risk allele might experience an intermediate phenotype (illustrating an additive model), except if APOL1 isoforms tends to preferentially form heterodimers, driving the number of deleterious homodimers down, limiting the toxicity and therefore maintaining cell homeostasis. Alternatively, individuals with one risk allele might also tolerate the increased toxicity (only 1/4 of damaging toxins in a dimer scenario vs. 1/2 in the monomer scenario) and maintain cell homeostasis. In the eventuality of a higher order APOL1 multimer (n-mer), the fraction of monotypic/damaging toxins would decrease (1/2n) as the number of APOL1 subunits required to form a functional toxin increases, thereby limiting the effect of the risk variants in individuals with one risk allele (Figure 3). These two models are not mutually exclusive and either would reconcile the recessive model of inheritance with the epidemiological data.
We recently provided evidence by size exclusion chromatography that APOL1 can multimerize in vitro. Western blotting and co-immunoprecipitation experiments from cell lysates further confirmed APOL1 multimerization (Dummer PD et al., 10th International Podocyte Conference, Freiburg, Germany, 2014; Kopp JB, personal communication). Whether APOL1 multimerization is important for APOL1 function and is involved in kidney injury remains to be formally demonstrated. Finally, this testable model contains several caveats and it is likely that the pathophysiologic reality is more complex than pictured in our schematic model, which should raise additional questions: there may be a balance between monomeric and multimeric forms, differences in assembly kinetics, protein abundance, localization, or binding affinities among the various APOL1 isoforms or between the various APOL1 isoforms and binding partner(s). By analogy with T.b. rhodesiense restriction, the G1 and G2 isoforms might alter the APOL1 renal function through different mechanisms (toxicity, membrane access and stabilization, and second factor binding).
Conclusions
Advances in understanding the mechanism by which APOL1 G1 and G2 variants cause renal injury have not kept pace with our rapidly expanding knowledge of the genetic epidemiology and disease associations of the APOL1 risk variants. There is suggestive evidence that APOL1 risk variants interacting with either genetic or environmental factors initiate renal programmed cell death and specific forms of kidney disease. Any mechanism for the pathophysiology underpinning APOL1-mediated disease will have to be consistent with a recessive mode of inheritance and the complementation of G1 and G2 in renal function despite apparent distinct trypanolytic mechanisms. Important clues for how APOL1 G1 and G2 proteins injure the renal cells may be gleaned from the mechanism of APOL1 trypanosome killing (destabilization of the lysosome membrane by pore formation controlled by the SRA binding) and from the function of proteins exhibiting a similar structural organization (multimerization). Deciphering the molecular mechanisms by which APOL1 damages kidney cells is essential for translating APOL1 genetic associations to effective preventive or therapeutic strategies and expanding the role for genetically informed medicine in the nephrology clinic.
Acknowledgments
We apologize to those authors whose work could not be included owing to space constraints. This work has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN26120080001E and by the Intramural Research Programs of Frederick National Laboratory, Center for Cancer Research, and NIDDK, NIH. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Footnotes
Disclosure. All the authors declared no competing interests.
References
- 1.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–5. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duchateau PN, Pullinger CR, Cho MH, et al. Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J Lipid Res. 2001;42(4):620–30. [PubMed] [Google Scholar]
- 3.Monajemi H, Fontijn RD, Pannekoek H, et al. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics. 2002;79(4):539–46. doi: 10.1006/geno.2002.6729. [DOI] [PubMed] [Google Scholar]
- 4.Page NM, Butlin DJ, Lomthaisong K, et al. The human apolipoprotein L gene cluster: identification, classification, and sites of distribution. Genomics. 2001;74(1):71–8. doi: 10.1006/geno.2001.6534. [DOI] [PubMed] [Google Scholar]
- 5.Vanhollebeke B, Pays E. The function of apolipoproteins L. Cell Mol Life Sci. 2006;63(17):1937–44. doi: 10.1007/s00018-006-6091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309(5733):469–72. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 7.Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422(6927):83–7. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 8.Albert TS, Duchateau PN, Deeb SS, et al. Apolipoprotein L-I is positively associated with hyperglycemia and plasma triglycerides in CAD patients with low HDL. J Lipid Res. 2005;46(3):469–74. doi: 10.1194/jlr.M400304-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Duchateau PN, Movsesyan I, Yamashita S, et al. Plasma apolipoprotein L concentrations correlate with plasma triglycerides and cholesterol levels in normolipidemic, hyperlipidemic, and diabetic subjects. J Lipid Res. 2000;41(8):1231–6. [PubMed] [Google Scholar]
- 10.Ma L, Shelness GS, Snipes JA, et al. Localization of APOL1 Protein and mRNA in the Human Kidney: Nondiseased Tissue, Primary Cells, and Immortalized Cell Lines. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2013091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lan X, Jhaveri A, Cheng K, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307(3):F326–36. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant. 2012;12(7):1924–8. doi: 10.1111/j.1600-6143.2012.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11(5):1025–30. doi: 10.1111/j.1600-6143.2011.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poelvoorde P, Vanhamme L, Van Den Abbeele J, et al. Distribution of apolipoprotein L-I and trypanosome lytic activity among primate sera. Mol Biochem Parasitol. 2004;134(1):155–7. doi: 10.1016/j.molbiopara.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Vanhollebeke B, Pays E. The trypanolytic factor of human serum: many ways to enter the parasite, a single way to kill. Mol Microbiol. 2010;76(4):806–14. doi: 10.1111/j.1365-2958.2010.07156.x. [DOI] [PubMed] [Google Scholar]
- 16.Molina-Portela Mdel P, Lugli EB, Recio-Pinto E, et al. Trypanosome lytic factor, a subclass of high-density lipoprotein, forms cation-selective pores in membranes. Mol Biochem Parasitol. 2005;144(2):218–26. doi: 10.1016/j.molbiopara.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Uzureau P, Uzureau S, Lecordier L, et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature. 2013;501(7467):430–4. doi: 10.1038/nature12516. [DOI] [PubMed] [Google Scholar]
- 18.Thomson R, Genovese G, Canon C, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci U S A. 2014;111(20):E2130–9. doi: 10.1073/pnas.1400699111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecordier L, Vanhollebeke B, Poelvoorde P, et al. C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog. 2009;5(12):e1000685. doi: 10.1371/journal.ppat.1000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nichols B, Jog P, Lee JH, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2014 doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22(11):2129–37. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horrevoets AJ, Fontijn RD, van Zonneveld AJ, et al. Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood. 1999;93(10):3418–31. [PubMed] [Google Scholar]
- 23.Sana TR, Janatpour MJ, Sathe M, et al. Microarray analysis of primary endothelial cells challenged with different inflammatory and immune cytokines. Cytokine. 2005;29(6):256–69. doi: 10.1016/j.cyto.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Taylor HE, Khatua AK, Popik W. The innate immune factor apolipoprotein L1 restricts HIV-1 infection. J Virol. 2014;88(1):592–603. doi: 10.1128/JVI.02828-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhaorigetu S, Wan G, Kaini R, et al. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4(8):1079–82. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samanovic M, Molina-Portela MP, Chessler AD, et al. Trypanosome lytic factor, an antimicrobial high-density lipoprotein, ameliorates Leishmania infection. PLoS Pathog. 2009;5(1):e1000276. doi: 10.1371/journal.ppat.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boasso A, Shearer GM. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin Immunol. 2008;126(3):235–42. doi: 10.1016/j.clim.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freedman BI, Skorecki K. Gene-Gene and Gene-Environment Interactions in Apolipoprotein L1 Gene-Associated Nephropathy. Clin J Am Soc Nephrol. 2014 doi: 10.2215/CJN.01330214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009;19(5):850–8. doi: 10.1101/gr.085647.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhaorigetu S, Yang Z, Toma I, et al. Apolipoprotein L6, induced in atherosclerotic lesions, promotes apoptosis and blocks Beclin 1-dependent autophagy in atherosclerotic cells. J Biol Chem. 2011;286(31):27389–98. doi: 10.1074/jbc.M110.210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu CA, Klopfer EI, Ray PE. Human apolipoprotein L1 (ApoL1) in cancer and chronic kidney disease. FEBS Lett. 2012;586(7):947–55. doi: 10.1016/j.febslet.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan G, Zhaorigetu S, Liu Z, et al. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283(31):21540–9. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galindo-Moreno J, Iurlaro R, El Mjiyad N, et al. Apolipoprotein L2 contains a BH3-like domain but it does not behave as a BH3-only protein. Cell Death Dis. 2014;5:e1275. doi: 10.1038/cddis.2014.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5(3):189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z, Lu H, Jiang Z, et al. Apolipoprotein l6, a novel proapoptotic Bcl-2 homology 3-only protein, induces mitochondria-mediated apoptosis in cancer cells. Mol Cancer Res. 2005;3(1):21–31. [PubMed] [Google Scholar]
- 36.Hartleben B, Godel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120(4):1084–96. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber TB, Edelstein CL, Hartleben B, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8(7):1009–31. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Periyasamy-Thandavan S, Jiang M, Schoenlein P, et al. Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297(2):F244–56. doi: 10.1152/ajprenal.00033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Periyasamy-Thandavan S, Jiang M, Wei Q, et al. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74(5):631–40. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Choi ME. Autophagy in kidney health and disease. Antioxid Redox Signal. 2014;20(3):519–37. doi: 10.1089/ars.2013.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomson R, Molina-Portela P, Mott H, et al. Hydrodynamic gene delivery of baboon trypanosome lytic factor eliminates both animal and human-infective African trypanosomes. Proc Natl Acad Sci U S A. 2009;106(46):19509–14. doi: 10.1073/pnas.0905669106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–69. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Limou S, Nelson GW, Kopp JB, et al. APOL1 Kidney Risk Alleles: Population Genetics and Disease Associations. Adv Chronic Kidney Dis. 2014;21(5):426–33. doi: 10.1053/j.ackd.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnstone DB, Shegokar V, Nihalani D, et al. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS One. 2012;7(12):e51546. doi: 10.1371/journal.pone.0051546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanhollebeke B, Truc P, Poelvoorde P, et al. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N Engl J Med. 2006;355(26):2752–6. doi: 10.1056/NEJMoa063265. [DOI] [PubMed] [Google Scholar]
- 46.Murphy JR. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins (Basel) 2011;3(3):294–308. doi: 10.3390/toxins3030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kent MS, Yim H, Murton JK, et al. Oligomerization of membrane-bound diphtheria toxin (CRM197) facilitates a transition to the open form and deep insertion. Biophys J. 2008;94(6):2115–27. doi: 10.1529/biophysj.107.113498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharpe JC, London E. Diphtheria toxin forms pores of different sizes depending on its concentration in membranes: probable relationship to oligomerization. J Membr Biol. 1999;171(3):209–21. doi: 10.1007/s002329900572. [DOI] [PubMed] [Google Scholar]
- 49.Steere B, Eisenberg D. Characterization of high-order diphtheria toxin oligomers. Biochemistry. 2000;39(51):15901–9. doi: 10.1021/bi0011678. [DOI] [PubMed] [Google Scholar]
- 50.Cheng YS, Hsia KC, Doudeva LG, et al. The crystal structure of the nuclease domain of colicin E7 suggests a mechanism for binding to double-stranded DNA by the H-N-H endonucleases. J Mol Biol. 2002;324(2):227–36. doi: 10.1016/s0022-2836(02)01092-6. [DOI] [PubMed] [Google Scholar]
- 51.Greig SL, Radjainia M, Mitra AK. Oligomeric structure of colicin ia channel in lipid bilayer membranes. J Biol Chem. 2009;284(24):16126–34. doi: 10.1074/jbc.M900292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steer BA, DiNardo AA, Merrill AR. Colicin E1 forms a dimer after urea-induced unfolding. Biochem J. 1999;340(Pt 3):631–8. [PMC free article] [PubMed] [Google Scholar]
- 53.Antonsson B, Montessuit S, Lauper S, et al. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345(Pt 2):271–8. [PMC free article] [PubMed] [Google Scholar]
- 54.Lovell JF, Billen LP, Bindner S, et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135(6):1074–84. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Peng J, Ding J, Tan C, et al. Oligomerization of membrane-bound Bcl-2 is involved in its pore formation induced by tBid. Apoptosis. 2009;14(10):1145–53. doi: 10.1007/s10495-009-0389-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peng J, Lapolla SM, Zhang Z, et al. The cytosolic domain of Bcl-2 oligomerizes to form pores in model mitochondrial outer membrane at acidic pH. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2009;26(3):631–7. [PMC free article] [PubMed] [Google Scholar]
- 57.Zha H, Aime-Sempe C, Sato T, et al. Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J Biol Chem. 1996;271(13):7440–4. doi: 10.1074/jbc.271.13.7440. [DOI] [PubMed] [Google Scholar]