

Abstract
Acute hypoxia depolarizes carotid body chemoreceptor (glomus) cells and elevates intracellular Ca2+ concentration ([Ca2+]i). Recent studies suggest that hydrogen sulfide (H2S) may serve as an oxygen sensor/signal in the carotid body during acute hypoxia. To further test such a role for H2S, we studied the effects of H2S on the activity of TASK channel and [Ca2+]i, which are considered important for mediating the glomus cell response to hypoxia. Like hypoxia, NaHS (a H2S donor) inhibited TASK activity and elevated [Ca2+]i. To inhibit the production of H2S, glomus cells were incubated (3 hr) with inhibitors of cystathionine-β-synthase and cystathionine-γ-lyase (DL-propargylglycine, aminooxyacetic acid, β-cyano-L-alanine; 0.3 mM). SF7 fluorescence was used to assess the level of H2S production. The inhibitors blocked L-cysteine- and hypoxia-induced elevation of SF7 fluorescence intensity. In cells treated with the inhibitors, hypoxia produced an inhibition of TASK activity and a rise in [Ca2+]i, similar in magnitude to those observed in control cells. L-cysteine produced no effect on TASK activity or [Ca2+]i and did not affect hypoxia-induced inhibition of TASK and elevation of [Ca2+]i. These findings suggest that under normal conditions, H2S is not a major signal in hypoxia-induced modulation of TASK channels and [Ca2+]i in isolated glomus cells.
Keywords: Hypoxia, Carotid body, Chemoreceptors, Hydrogen sulfide, L-cysteine
1. Introduction
Hypoxia depolarizes carotid body glomus cells by inhibiting the outward K+ current and elevates [Ca2+]i by opening voltage-gated Ca2+ channels. The rise in [Ca2+]i increases the secretory event leading to augmented activity of the carotid sinus afferent nerve (Lopez-Barneo et al., 2004; Pardal et al., 2000). Although this general scheme of events is well accepted, the intracellular O2 sensor and associated signals that mediate the hypoxia-induced excitation of glomus cells via modulation of ion channels are still not well defined. A number of recent studies have focused on the potential role of H2S as an O2 sensor/signal in the response of the carotid body (CB) to hypoxia. So far, evidence both in support of and against a role for H2S has been presented, and the issue remains unresolved.
In those studies with positive findings, H2S increased the carotid sinus afferent nerve activity in carotid sinus nerve-CB preparation and increased [Ca2+]i in isolated glomus cells (Li et al., 2010; Makarenko et al., 2012; Peng et al., 2010). In these studies, inhibitors of cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE) that normally produce H2S in the cell markedly reduced the chemoreceptor afferent nerve activity as well as the rise in [Ca2+]i in response to hypoxia. Glomus cells from mice with deletion of the CSE gene (CSE−/−) also showed a strongly reduced catecholamine secretion in response to hypoxia, compared with cells from wild type mice (Peng et al., 2010). CSE−/− mice also exhibited an impaired ventilatory response to hypoxia (Peng et al., 2010). Together, these findings support the hypothesis that H2S mediates the ventilatory response to hypoxia by increasing glomus cell [Ca2+]i, the secretion of transmitters and the chemosensory response of the CB.
Arguing against the hypothesis that H2S is a mediator of the glomus cell response to hypoxia are findings in which NaHS or Na2S (H2S donors) inhibited the transmitter release from cat CB (Fitzgerald et al., 2011), and reduced mitochondrial function, as assessed by NADH autofluorescence and measurement of intracellular [Mg2+] that was taken as an index of ATP concentration (Buckler, 2012). Any agent that inhibits glomus cell mitochondrial oxidative phosphorylation would be expected to produce effects similar to those of hypoxia. Therefore, it was argued that the effect of exogenously applied H2S on glomus cells was simply due to an effect on mitochondrial function similar to that produced by hypoxia (Buckler, 2012). In another study, an indirect sequestration of H2S using methemoglobin (produced by injection of sodium nitrite) did not prevent hypoxia-induced hyperventilation (Haouzi et al., 2011a; Haouzi et al., 2011b). These findings therefore do not support a role for H2S as an intracellular signal that mediates the CB glomus cell response to hypoxia.
We hypothesized that if endogenous H2S were a true mediator of the glomus cell response to hypoxia, it would cause inhibition of K+ channels known to be involved in hypoxia-induced depolarization and elevate [Ca2+]i in isolated glomus cells. Such effects of H2S on TASK and BK have been reported recently (Buckler, 2012; Telezhkin et al., 2010). More importantly, the hypoxia-induced inhibition of the K+ channels and elevation of [Ca2+]i should be blocked or strongly reduced when the endogenous H2S production is eliminated. Reduction of hypoxia-induced rise in [Ca2+]i by inhibitors of CSE and in CSE−/− mice has been reported recently (Makarenko et al., 2012), but the effect of inhibitors of CSE and CBS on hypoxia-induced inhibition of K+ channels remains to be determined. We believe that these experiments are crucial tests to show whether H2S is indeed an intracellular signal mediating the hypoxia response in glomus cells. In this study, we tested the effect of inhibitors of CSE and CBS on hypoxia-induced inhibition of TASK channel, and measured changes in [Ca2+]i to revisit the earlier finding that the inhibitors of these enzymes reduce the [Ca2+]i response to hypoxia. We also tested the effect of elevating endogenous production of H2S using L-cysteine on TASK and [Ca2+]i. To our surprise, our results showed that L-cysteine or inhibitors of CSE and CBS failed to block the hypoxia-induced inhibition of TASK channel and the rise in [Ca2+]i. Our findings therefore do not support the role of H2S as an O2 sensor/signal in hypoxia-induced excitation of rat glomus cells.
2. Methods
2.1. Cell isolation
Rats (postnatal 18–24 day; Sprague-Dawley) were anesthetized with isoflurane and used according to the animal protocols approved by the Animal Care and Use Committees of Rosalind Franklin University and University of Arkansas for Medical Sciences. The carotid bodies were removed and placed in ice-cold low-Ca2+, low-Mg2+ phosphate buffered saline solution (low Ca2+/Mg2+-PBS: 137 mM NaCl, 2.8 mM KCl, 2 mM KH2PO4, 0.07 mM CaCl2, 0.05 mM MgCl2, pH 7.4). Each carotid body was cut into 2–3 pieces and placed in a solution containing trypsin (400 mg/ml) and collagenase (400 mg/ml) in low Ca2+/Mg2+-PBS and incubated at 37°C for 20–25 min. Carotid bodies were gently triturated using a fire polished Pasteur pipette to mechanically dissociate the cells. Growth medium (Ham’s F-12, 10% fetal bovine serum, 23 mM glucose, 2 mM L-glutamine, 10K units penicillin/streptomycin, and 300 mg/ml insulin) was added to stop enzyme activity. After brief trituration, the solution containing the digested carotid bodies was centrifuged for 4 min at ~6000 rpm (~2000 x g) using a microcentrifuge. The supernatant was removed and warm growth medium added to gently resuspend the pellet. Suspended cells were placed on glass coverslips coated with poly-L-lysine, and incubated at 37°C in a humidified atmosphere of 95% air-5% CO2. Cells were used 3 hr later.
2.2. Electrophysiological studies
Electrophysiological recording was performed using a patch clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA). Patches were formed using borosilicate glass pipettes with 3–5 megaohm tip resistance. The pipette solution contained (mM) 150 KCl, 1 MgCl2, 5 EGTA, 10 glucose and 10 HEPES (pH 7.3), and the bath perfusion solution contained (mM) 117 NaCl, 5 KCl, 23 NaHCO3, 1 CaCl2, 1 MgCl2 and 11 glucose (pH 7.3). Channel current was filtered at 3 kHz using 8-pole Bessel filter (−3 dB; Frequency Devices, Haverhill, MA) and transferred to a computer using the Digidata 1320 interface at a sampling rate of 20 kHz. Single-channel currents were analyzed with the pCLAMP program (Version 10). Channel openings were analyzed to obtain channel activity (NPo, where N is the number of channels in the patch, and Po is the open probability of a channel). NPo was determined from 15–30 s of current recordings. Because glomus cells express both ~16-pS (TASK-1) and ~35-pS (TASK-3 and TASK-1/3) channels, analysis was done to detect all three isoforms by setting the open levels as multiples of ~16-pS channel. Single-channel current tracings shown in figures were filtered at 1 kHz. All electrophysiological experiments were performed at ~35°C.
2.3. [Ca2+]i measurement
[Ca2+]i was measured by quantitative fluorescence imaging using the calcium-sensitive dye fura-2. Cells plated on a coverslip were incubated with 4 μM fura-2 acetoxymethyl ester (fura-2 AM; Molecular Probes) for 30 min at 37°C. Fura-2 fluorescence emission was measured at 510 nm in response to alternating excitation at 340 and 380 nm. Images were acquired and stored using a NIKON Eclipse TE300 microscope (with 40x oil immersion objective) and CCD (CoolSNAP HQ2) camera under computer control (MetaFluor: Molecular Devices). For each coverslip, the background light levels were determined and subtracted from each image before measurement of the fluorescence intensity ratio. [Ca2+]i was determined using the 340 nm/380 nm fluorescence ratio as described previously (Grynkiewicz et al., 1985). Calibration was performed using cell-free solutions. The perfusion solution used for [Ca2+] measurements contained (mM): 118 NaCl, 23 NaHCO3, 3 KCl, 2 KH2PO4, 1 MgCl2, 1.2 CaCl2, 11 glucose. The temperature of the perusion solution at the recording chamber was 35°C.
2.3. SF7 fluorescence recording
Endogenous production of intracellular [H2S] was estimated using SF7-AM (sulfide fluor-7-acetoxymethyl ester; gift from Dr. Christopher Chang, University of California, Berkeley), a recently developed fluorescence probe for H2S (Lin et al., 2013). Cells plated on a glass coverslip were incubated with SF7-AM (2.5 μM) for 10 min at 37°C. Excitation and emission wavelengths were 490 nm and 525 nm, respectively. Images were acquired and stored using a Nikon Eclipse TE300 microscope (with 40x oil immersion objective) and CCD (CoolSNAP HQ2) camera under computer control (NIS Element, Nikon). The data are shown as fluorescence unit. The perfusion solution (35°C) used for measurement of SF7 signal contained (mM): 118 NaCl, 23 NaHCO3, 2 KH2PO4, 3 KCl, 1 MgCl2, 1.2 CaCl2, 11 glucose.
2.4. Hypoxia studies
Cell-attached patches were formed on glomus cells and perfused with a bicarbonate-buffered solution containing (mM) 117 NaCl, 5 KCl, 23 NaHCO3, 1 MgCl2 and 11 glucose, and bubbled with 5% CO2/95% air mixture (normoxia) for ~60 min. After steady state channel activity was obtained, the perfusion solution was switched to solution bubbled vigorously (for at least 60 min at 37°C prior) with 5% CO2/95% N2 mixture (hypoxia) for desired periods of time. The pipette solution contained (mM) 150 KCl, 1 MgCl2, 5 EGTA, 10 glucose and 10 HEPES (pH 7.3). The temperature of the perfusion solutions was kept at ~35°C. O2 pressure of the solutions was determined using an oxygen meter (ISO2, WPI, Sarasota, USA) that was calibrated to 0% with solution gassed with pure nitrogen for 60 min and to 21% with solution gassed with air for 60 min at 35°C. The O2 partial pressures in the perfusion solution inside the perfusion chambers used for single channel recording and fluorescence measurements were ~8 mmHg (~1.0% O2) and ~4 mmHg (~0.5% O2), respectively.
2.5. Statistical analysis
Student’s t-test (for comparison of two sets of data) and one-way analysis of variance (for comparison of three data sets) were used. Data were analyzed using PRISM software and represented as mean±SE, unless indicated otherwise. Post hoc testing was based on unpaired t-test with Bonferroni correction. Significance level was set at p<0.05.
2.6. Materials
DL-propargylglycine, aminooxyacetic acid, β-cyano-L-alanine, L-cysteine, and sodium hydrogen sulfide were purchased from Sigma-Aldrich Co.
3. Results
3.1. Hypoxia and NaHS inhibit TASK activity and depolarize glomus cells
We first repeated the earlier experiment to confirm that hypoxia and H2S inhibit TASK activity and depolarize glomus cells. As reported earlier (Buckler, 2007; Kim et al., 2009), cell-attached patches with 140 mM KCl in the pipette solution and physiological bath solution containing 5 mM KCl showed opening of mainly TASK channels that were basally active (Fig. 1A: tracing a). TASK was identified by the main single channel conductance level of ~35-pS and short mean open time of ~0.5 ms. Single TASK channel current reversed at the pipette potential of ~ −60 mV, suggesting that the resting Em of glomus cells is probably close to this negative potential, assuming that intracellular [K+] in glomus cells is high (~140 mM).
Figure 1. Inhibition of TASK activity by hypoxia and NaHS in glomus cells.

A. Current tracings show TASK channel openings in a cell-attached patch perfused with normoxic and hypoxic solutions. Hypoxia produced a rapid and reversible inhibition of TASK activity, as illustrated by expanded current tracings (indicated by lower case letters). Dotted lines indicate the closed state of the channel.
B. Current tracings show TASK channel openings in a cell-attached patch in response to NaHS. NaHS produced a rapid and reversible inhibition of TASK activity. Expanded current tracings are also shown, as indicated by lower case letters.
C. The graph shows a plot of TASK activity as a function of [NaHS]. The data points were fitted to an Hill equation of the form: y=((1−Ao)/(1+(x/xo)n))+Ao, where Ao is determined to be 0.17, xo is 22 μM (K1/2) and n is 1.62. Each point is mean±SE of 5 determinations.
D. Current tracing shows a lack of response to 100 μM NaHS in an inside-out patch in the presence of 4 mM ATP in the bath solution.
E. Summary data from all experiments are shown. Each bar is the mean±SE of 7–8 determinations obtained from three cell preparations. Significant difference (p<0.05) is indicated by an asterisk(*).
Switch of the bath perfusion from a normoxic (21% O2) to a hypoxic solution (0% O2) quickly and reversibly reduced TASK activity and the amplitude of single channels (Fig. 1A: tracings a and b). The magnitude of reduction of TASK single channel amplitude is linearly dependent on cell membrane potential such that a decrease in amplitude by 50% indicates a depolarization of ~30 mV (Kim et al., 2009). From this relationship, we determined that hypoxia produced a depolarization of 28±3 mV in this set of experiments (n=4). Perfusion of cell-attached patches with 100 μM NaHS also produced a rapid and reversible inhibition of TASK activity (Fig. 1B; tracings a and b). Half-maximal inhibition of TASK was observed at ~22 μM NaHS (Fig. 1C). NaHS (100 μM) also caused cell depolarization of ~36 mV from the resting Em. In inside-out patches, TASK activity quickly runs down and can be recovered by addition of ATP (Varas et al., 2007; Williams and Buckler, 2004). In the presence of 4 mM ATP in the bath (cytosolic) solution, 100 μM NaHS did not affect TASK activity (Fig. 1D). These results summarized in Fig. 1E show that both hypoxia and NaHS are strong inhibitors of TASK activity as reported previously, and that the inhibition of TASK activity by NaHS in intact glomus cells occurs indirectly via an intracellular target.
3.2. Inhibitors of CBS and CSE do not affect hypoxia-induced inhibition of TASK activity
An important test of the role of H2S as an intracellular physiologic mediator of the glomus cell response to hypoxia is to show that blocking endogenous H2S generation eliminates or reduces the glomus cell response to hypoxia. To test such a role for H2S, glomus cells were incubated for 10 min at 37°C with 0.3 mM DL-propargylglycine (PPG) and 0.3 mM amino-oxyacetic acid (AOAA) that have been shown to inhibit CSE and CBS, respectively (Asimakopoulou et al., 2013). Cell-attached patches were then formed in the continuous presence of these inhibitors, and TASK activity recorded before and after perfusion with the hypoxic bath solution. Contrary to our expectation, hypoxia inhibited TASK activity to similar extent in control (0.1% DMSO) and PPG/AOAA-treated glomus cells (Fig. 2B). In glomus cells incubated with the inhibitors, hypoxia also reduced the single channel amplitude of TASK. The calculated shift in resting Em (30±3 mV; n=5) produced by hypoxia was not different from those observed in cells not treated with the inhibitors.
Figure 2. Effect of PPG and AOAA on hypoxia-induced changes in TASK activity.
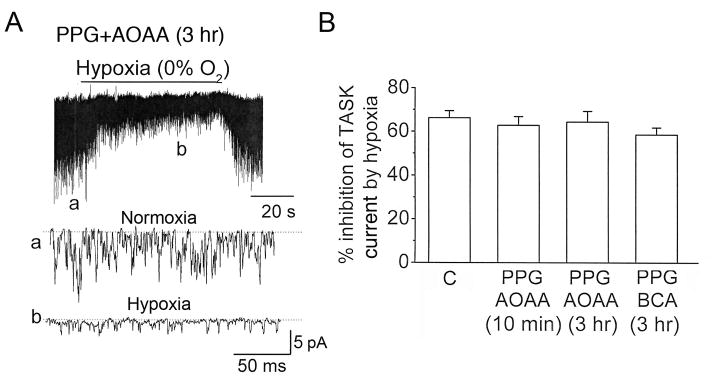
A. Glomus cells were incubated with PPG (0.3 mM) and AOAA (0.3 mM) for 3 hr. Current tracings show TASK channel openings in a cell-attached patch perfused with normoxic and hypoxic solutions. Hypoxia produced a rapid and reversible inhibition of TASK activity, as illustrated by expanded current tracings.
B. Summary results obtained using PPG and AOAA as well as PPG and BCA (0.3 mM) are shown. Cells were pre-incubated at 37°C with either 0.1% DMSO (labeled as C) or with the inhibitors for 10 min or ~3 hr. The effect of hypoxia on TASK activity was then determined from cell-attached patches. Bars represent channel activity percent inhibition of TASK activity hypoxia for each group. Each data point is the mean±SE of 6 determinations obtained from three cell preparations. There were no significant differences between any two groups (p>0.05).
When glomus cells were pre-incubated with PPG and AOAA for a longer time period (~3 hr), the inhibition of TASK activity produced by hypoxia was also not significantly different from those obtained from control glomus cells (Fig. 2A and 2B). The degree of depolarization (28±3 mV; n=4), as judged by the reduction of TASK single channel amplitude, was also not significantly different from that observed in control cells (32±4 mV; n=4). Pre-incubation of glomus cells with PPG and β-cyano-L-alanine (another inhibitor of CSE) also failed to blunt the inhibition of TASK activity by hypoxia (Fig. 2B). These results suggest that endogenously generated H2S may not mediate the hypoxia-induced inhibition of TASK activity and depolarization of glomus cells.
3.3. PPG and AOAA inhibit endogenous H2S production in glomus cells
PPG and AOAA have been used extensively to inhibit CBS and CSE in many studies. However, we felt it necessary to show such inhibition directly in our CB glomus cell preparations to be certain that H2S production was actually blocked. We used sulfide fluor7-AM (SF7-AM), a recently developed fluorescent probe for for detection of H2S (Lin et al., 2013). To show that PPG and AOAA are effective inhibitors of H2S production in glomus cells, L-cysteine (10 mM) was added to the perfusion solution while recording the fluorescence intensity (Fig. 3A). L-cysteine is known to stimulate the production of H2S in tissues by serving as the substrate for CSE and CBS (Wang, 2012). Under control perfusion conditions, the basal level of fluorescence intensity tended to increase slowly in a linear fashion, which we suspect is due to the properties of the dye itself. After several minutes of perfusion with L-cysteine, the fluorescence intensity started to increase more rapidly and reached a steady state level after ~15 min (Fig. 3A). When L-cysteine was applied to cells that were pre-incubated with PPG and AOAA (0.3 mM each) for 3 hr, the increase in fluorescence intensity was markedly reduced (Fig. 3B). Fig. 3C shows the summary of these experiments.
Figure 3. SF7 fluorescence signal recording from isolated glomus cells.

A. Averaged fluorescence intensity from glomus cells is shown. After three hours incubation with no CSE or CBS inhibitors (incubation time control for data shown in Figure 3B), L-cysteine (10 mM) produced a clearly identifiable increase in fluorescence signal. Short unfilled parts of the tracings in some tracings are due to brief interruptions by a timing device (vertical dotted lines).
B. Glomus cells were incubated in culture medium containing PPG (0.3 mM) and AOAA (0.3 mM) for 3 hr. In these cells, L-cysteine (10 mM) produced a markedly reduced increase in fluorescence signal.
C. Summary bar graph shows the effect of L-cysteine on SF7 fluorescence. The first bar (L-cysteine alone) is the mean±SE of 41 glomus cells combined from two experiments (12 CBs). The second bar (L-cysteine+inhibitors) is the mean±SE of 16 cells from one experiment (6 CBs). Significant difference (p<0.001) is indicated by an asterisk(*).
D. Averaged fluorescence intensity from glomus cells in normoxia and hypoxia is shown. Hypoxia increased SF7 fluorescence signal. In the second hypoxia treatment, the perfusion solution contained 0.5 mM Na2S2O4 to make the solution anoxic.
E. In cells pre-incubated in culture medium containing PPG (0.3 mM) and AOAA (0.3 mM) for 3 hr, hypoxia produced only a very small increase in fluorescence signal.
F. Summary bar graph shows the composite data of the effect of hypoxia on SF7 fluorescence. Each bar is the mean±SE of 27–28 cells prepared from 12 CBs (two separate experiments). Significant difference (p<0.001) is indicated by an asterisk(*).
To test whether PPG and AOAA also block hypoxia-induced elevation of H2S production, cells were incubated with the inhibitors for 3 hr and then perfused with normoxic (21% O2) and hypoxic solutions (0% O2). In control untreated cells, perfusion with the hypoxic solution increased the fluorescence intensity that peaked at ~7 min in this experiment (Fig. 3D). The fluorescence intensity decreased slowly when cells were perfused with a normoxic solution. Perfusion of cells with an anoxic solution produced by adding 0.5 mM Na2S2O4 to the hypoxic solution again increased the fluorescence intensity. In cells that were pre-incubated with PPG and AOAA for 3 hr, the hypoxic solution showed only a small increase in fluorescence signal (Fig. 3E). Fig. 3F shows the summary of results on the effects of hypoxia on SF7 fluorescence. These results show that the inhibitors of CSE and CBS used here strongly reduce the production of H2S in glomus cells, and are consistent with earlier studies showing that PPG and AOAA inhibit H2S production in different cell types (Fernandes et al., 2014; Porteus et al., 2014; Prieto-Lloret et al., 2014).
3.4. Inhibitors of CBS and CSE do not inhibit hypoxia-induced increase in [Ca2+]i
Another experiment to show that H2S mediates the glomus cell response to hypoxia is to measure [Ca2+]i, which is well known to be elevated by hypoxia. As shown previously, extracellular perfusion of glomus cells with solution containing high KCl (20 mM), low O2 tension, or NaHS elevated [Ca2+]i (Fig. 4A). Each stimulus produced a rapid and reversible increase in [Ca2+]i. To test the hypothesis that H2S mediates the hypoxia-induced elevation of [Ca2+]i, we again used PPG and AOAA to block the production of H2S in glomus cells before challenging the cells with a hypoxic solution. In cells pre-incubated with PPG and AOAA (0.3 mM each) for 3 hr, hypoxia produced an increase in [Ca2+]i similar in magnitude to those observed in control cells (Fig. 4B and D). When cells were incubated with 0.3 mM PPG and 0.3 mM BCA (β-cyano-L-alanine), another combination of inhibitors that are known to block CSE, hypoxia also elicited a rapid increase in [Ca2+]i, similar to those observed in control cells (Fig. 4C and D). The hypoxia-induced elevation of [Ca2+]i following washout of the inhibitors was also similar to those obtained in control and inhibitor-treated cells (Fig. 4D). Averaged data from three separate experiments showed that the degree of [Ca2+]i rise was similar in the presence and absence of the inhibitors. These results show that even after ~3 hr of incubation with the inhibitors when the endogenous production of H2S was strongly reduced (Fig. 3), the glomus cell response to hypoxia was essentially unchanged. These results suggest that H2S is not a major signal for the glomus cell response to hypoxia.
Figure 4. Inhibitors of CSE and CBS do not reduce the hypoxia-induced elevation of [Ca2+]i in glomus cells.

A. Changes in [Ca2+]i were recorded from glomus cells in response to 20 mM KCl, hypoxia (0% O2), 0.1 mM NaHS and 1 mM NaHS. Each stimulus produced a rapid and reversible increase in [Ca2+]i.
B. Changes in [Ca2+]i were recorded from cells pre-incubated with PPG (0.3 mM) and AOAA (0.3 mM) for 3 hr. Hypoxia elicited a rapid and reversible increase in [Ca2+]i that was similar in magnitude to those observed in control cells. After washout of PPG and AOAA for ~10 min, hypoxia elicited a [Ca2+]i rise of similar magnitude.
C. Same experiment as in B except that the cells were pre-incubated with PPG and BCA (0.3 mM each) for ~1 hr. In these cells, hypoxia also elicited a [Ca2+]i rise similar to that observed in control cells.
D. Summary of glomus cell [Ca2+]i responses to hypoxia during perfusion with inhibitors (indicated by arrows) and after washout (labeled W) compared to control (labeled C). No significant difference (p>0.05) in [Ca2+]i rise was present with and without the pre-incubation of cells with the drugs. Each bar is the mean±SE of 24 cells from three different cell preparations (total of 18 CBs).
3.5. Studies using L-cysteine
L-cysteine has been used to increase the endogenous production of H2S in a variety of cell types (Elsey et al., 2010; Kaneko et al., 2006; Olson et al., 2008). To test the effect of L-cysteine on TASK activity, glomus cells were incubated in perfusion solution containing 10 mM L-cysteine, and cell-attached patches were formed in the continuous presence of L-cysteine. TASK activity monitored in normoxic and hypoxic solutions at various time points following incubation with L-cysteine. Fig. 5A shows a current tracing from a glomus cell incubated with L-cysteine for 23 min, and expanded current tracings are also shown. In Fig. 5B, each point represents TASK activity in a cell-attached patch in normoxia after some time period of incubation with L-cysteine. TASK activity in cell-attached patches of control cells without L-cysteine treatment in normoxia was 0.32±0.03 (n=6) and remained unchanged during the 27 min of perfusion with L-cysteine (Fig. 5B). In 12 cells incubated with L-cysteine for 15–27 min, the mean TASK activity was 0.31±0.02 (n=12). L-cysteine (15–27 min treatment) also did not affect the mean TASK single channel amplitude (2.0±0.1 pA vs. 1.9±0.1 pA; p<0.05; n=5), indicating that the cell membrane potential remained unchanged. After exposure to L-cysteine, some patches were further exposed to a hypoxic solution. In these patches, hypoxia produced a 65% reduction of TASK activity (Fig. 5C), similar to that observed in control cells that were not exposed to L-cysteine (see Fig. 2). Thus, L-cysteine produced no significant effect on TASK activity at least for the duration of the experiment, and did not affect the degree of inhibition of TASK activity by hypoxia. These results suggest that endogenously produced H2S is not sufficiently high in concentration to inhibit TASK in intact isolated glomus cells.
Figure 5. Effects of L-cysteine and hypoxia on TASK and [Ca2+]i.

A. Glomus cell was superfused with solution containing 10 mM L-cysteine. At various time points after the perfusion with L-cysteine, cell-attached patches were formed and TASK recorded in normoxia and hypoxia. The tracing shown was obtained from a cell superfused with L-cysteine for 23 min.
B. TASK activities in cell-attached patches are plotted as a function of time after application of 10 mM L-cysteine. Each point represents TASK activity from one cell-attached patch. The mean TASK activity from cells incubated with L-cysteine for 15–27 min were 0.31±0.02 (n=12), compared to 0.32±0.03 in untreated control cells (n=6).
C. Bar graph shows averaged TASK activity in normoxia and hypoxia in cells incubated with L-cysteine for 15–25 min. Each bar is the mean±SE of 7 values. Significance (p<0.05) is denoted by asterisk(*).
D. Changes in [Ca2+]i in response to 20 mM L-cysteine are shown. After ~5 min washout of L-cysteine, hypoxic solution containing L-cysteine elicited a strong increase in [Ca2+]i. An averaged tracing from 12 cells from one experiment (6 CBs) is shown.
E. Same experiment as in B except that the effect of hypoxia was tested without washout L-cysteine. An averaged tracing from 12 cells from one experiment (6 CBs) is shown.
We also tested the effect of exogenous application of L-cysteine on [Ca2+]i in isolated glomus cells. During the ~15 min of perfusion of isolated glomus cells with L-cysteine, a small increase in basal [Ca2+]i was present (53±5 to 75±11 nM; Fig. 5D). After ~6 min washout of L-cysteine, application of hypoxia and L-cysteine elicited a strong increase in [Ca2+]i to 425±25 nM (Fig. 5D). In the continuous presence of L-cysteine, hypoxia also produced a similarly large increase in [Ca2+]i to 414±42 nM (Fig. 5E). Because a high concentration of L-cysteine is likely to produce not only H2S but also other sulfur-containing compounds, the interpretation of the results is not straightforward. Nevertheless, these results are consistent with the notion that H2S is not acting as an O2 sensor or a signal for hypoxia-induced excitation of glomus cells.
4. Discussion
The role of H2S as O2 sensor and signal in hypoxia-induced excitation of CB glomus cells is highly controversial. Inhibition of H2S-producing enzymes (CBS and CSE) or deleting the gene for CSE has been reported to markedly reduce the hypoxia-induced increase in ventilatory response, carotid sinus afferent nerve activity, transmitter secretion and [Ca2+]i rise in glomus cell (Li et al., 2010; Makarenko et al., 2012; Peng et al., 2010). Based on both experimental results and theoretical considerations, however, several groups of investigators have rejected the idea that H2S is a mediator of the glomus cell hypoxia response (Buckler, 2012; Fitzgerald et al., 2011; Haouzi et al., 2011a). To further examine the role of H2S, we reduced the endogenous production of H2S using inhibitors of CSE and CBS, and tested the effect of hypoxia on two events associated with the hypoxia-induced excitation of glomus cells: TASK channel inhibition and elevation of [Ca2+]i. Our findings on TASK channel and [Ca2+]i do not support the role of H2S as a major O2 sensor or signal that mediates the CB response to hypoxia.
4.1. Evidence for and against a role for H2S as the hypoxia-induced signal in the CB
In support of a role for H2S, studies reported that inhibitors of CBS and/or CSE or deletion of the CSE gene strongly reduced the chemosensory response to hypoxia (Li et al., 2010; Peng et al., 2010) and reduced hypoxia-induced elevation of [Ca2+]i (Makarenko et al., 2012). The reduction of ventilatory response to hypoxia by inhibitors of CBS and CSE (PPG and AOAA) was also recently observed in zebrafish (Porteus et al., 2014). One issue with the studies described above is that it is unclear which enzyme, CSE or CBS, is important for the response of the CB to hypoxia. Both CSE and CBS are expressed in the CB glomus cells, as judged by RT-PCR and immunochemistry (Telezhkin et al., 2010). In one study, an inhibitor of CSE (PPG) alone had no effect on the chemosensory response, whereas inhibitors of CBS (AOAA and hydroxylamine) markedly decreased it (Li et al., 2010). On the other hand, CSE was the main enzyme responsible for H2S production in hypoxia and PPG strongly reduced the chemosensory response (Peng et al., 2010). This discrepancy in the role of CBS and CSE is disturbing, as it is unlikely that both of these conflicting findings are correct. Both studies use mice preparations, and therefore species difference cannot explain the findings, unless different strains of mice show different hypoxia transduction mechanisms (Otsubo et al., 2011).
Although studies showing that hypoxia elevates [H2S] in the CB and that hypoxia-induced increase in chemosensory response and ventilation is markedly diminished in CSE−/− mice are very compelling, several issues question the validity of the H2S/O2 sensor model. In general, the arguments are based on the biochemical properties of H2S itself as well as on experimental results that disqualify H2S as the sole mediator of the glomus cell response to hypoxia. H2S is a potent inhibitor of cytochrome C oxidase and therefore probably activates all the signaling pathways identical to those activated by hypoxia and mitochondrial inhibitors when applied exogenously. The problem lies in the “nearly toxic” micromolar concentrations of H2S required to elicit the ventilatory and chemosensory response. Micromolar (10–300 μM) levels of H2S are required to elevate [Ca2+]i in glomus cells, increase chemosensory activity and inhibit K+ channels such as TASK and BK (Li et al., 2010; Telezhkin et al., 2010). It has been argued that such high [H2S] would not be achieved in vivo because it is too toxic for the organism, and that only nanomolar amounts are present in cells for their signaling requirements (Haouzi et al., 2011b). Our studies using L-cysteine also support this view. An efficient biochemical pathway for degradation of H2S as well as high solubility of H2S in blood are believed to ensure that only non-toxic levels of H2S exist in cells. Unfortunately, the physiological levels of H2S in different cellular compartments are not yet known to settle the issue of how high [H2S] truly is in native tissues.
In the cat CB, H2S was found to inhibit transmitter secretion (ATP and ACh), rather than augment it (Fitzgerald et al., 2011). This finding is rather surprising because H2S elevates glomus cell [Ca2+]i and therefore is expected to stimulate transmitter secretion. As discussed by the authors, H2S at the concentration used (5–100 μM) may be activating ATP-sensitive K+ channels to hyperpolarize the cells and limit transmitter secretion. In another study, sequestration of plasma H2S using methemoglobin did not block hypoxia-induced hyperventilation; however, it is unclear how much H2S was actually removed from the CB (Haouzi et al., 2011a). In the lung, H2S has been proposed to mediate the hypoxic pulmonary vasoconstriction (Olson, 2008; Olson et al., 2006). A more recent study showed, however, that PAG and AOAA failed to inhibit the hypoxic pulmonary vasoconstriction (Prieto-Lloret et al., 2014); in this study, PAG and AOAA strongly antagonized the release of sulfide from pulmonary arteries, as determined by amperometric methods. Thus, evidence both for and against a role for H2S in hypoxia-induced excitation in the CB have been presented and the controversy remains unresolved.
4.2. Increased endogenous production of H2S is not associated with glomus cell response to hypoxia
To better understand the role of H2S, we felt that it would be important to study the effect of hypoxia on TASK, Em and [Ca2+] in glomus cells when the production of H2S is blocked. In our study, hypoxia still caused a strong inhibition of TASK even after the endogenous production of H2S was strongly blocked with PPG and AOAA. Experiments using PPG and AOAA also showed that an increased endogenous production of H2S by hypoxia was not necessary for hypoxia-induced depolarization and elevation of [Ca2+]i. These findings indicate that hypoxia uses a signaling pathway that may not involve an increase in [H2S] to cause excitation of glomus cells. Because L-cysteine did not mimic the effect of hypoxia on [Ca2+]i, it seems most likely that the endogenous production of H2S produced by hypoxia is not sufficiently high in concentration to inhibit TASK and elevate [Ca2+]i. This is consistent with the findings of an earlier study in which 100 μM L-cysteine did not stimulate the CB sensory nerve activity and also did not enhance hypoxia-induced increase in nerve activity, despite the increased H2S level measured biochemically (Makarenko et al., 2012; Peng et al., 2010).
Although all inhibitors of CBS and CSE used here are non-specific, they strongly reduced the production of H2S in glomus cells, based on SF7 fluorescent measurements. In a mouse macrophage cell line (Raw), pre-incubation with PPG and AOAA for 1 hr also markedly reduced SF7 fluorescent intensity (Carl White, personal communication). Sulfidefluors have been synthesized to help detect H2S in cells (Lin and Chang, 2012; Lippert et al., 2011), and the latest generation of these compounds (SF5 and SF7) are able to detect H2S levels in the >250 nM range. SF7, the most advanced member of sulfidefluors, has been used successfully to detect vascular endothelial growth factor-dependent production of H2S in human umbilical vein endothelial cells (Lin et al., 2013). Although we are currently unable to continue to use SF7-AM to further study H2S production due to limited availability of the dye, our assessment of H2S production using SF7-AM so far is in excellent agreement with the well-known inhibitory actions of PPG, AOAA and BCA on CSE and CBS reported in many studies. In some of our experiments, we were not able to perform an experiment multiple times due to the unavailability of SF7-AM, but all of the data on TASK and [Ca2+]i measurements were consistent, further supporting our view that H2S is not the endogenous hypoxic signal for the excitation of isolated glomus cells.
4.3. Limitations of the study using isolated glomus cells
An important limitation of using isolated clustered glomus cells is that the results obtained may not represent the physiological and pathophysiological processes that occur in vivo. The CB contains many different types of cells, including cells of the vasculature where H2S is generated locally and regulates the vascular tone (Dombkowski et al., 2006; Morikawa et al., 2012). H2S production in the vasculature is elevated by hypoxia and this may affect the function of glomus cells as the gaseous H2S would diffuse within the CB. Thus, inhibitors of CBS and CSE may produce different levels of chemoreceptor activity in intact CB and isolated glomus cells. In future studies, it would be important to compare the effects of hypoxia as well as inhibitors of CBS and CSE on the production of H2S, potentially using SF7 in whole CB and isolated glomus cells.
A potential issue in our study relative to others may be the use of adolescent rats in our experiments. Many studies testing the role of H2S in carotid body O2 sensing used older aged rats ranging from several months to adult. Our study used rats that were 18–24 days of age. Numerous laboratories over the last two decades have examined the time course of postnatal maturation of carotid body function, mostly by measuring the neural response (carotid sinus nerve recordings) or the CB glomus cell [Ca2+]i response to hypoxia (Carroll and Kim, 2013). All of these studies agree that, for the rat, CB function has matured to adult levels by ~14 days. Therefore, from a CB O2-sensing development perspective, our studies done in rats 18–24 days should be comparable to studies performed in several month old and adult rats.
4.4. Pathophysiological role of H2S
In CB glomus cells, high [H2S] inhibits TASK, causes depolarization and elevates [Ca2+]i, which are events similar to those produced by hypoxia. Such effects of H2S on glomus cell function may have pathophysiological relevance if the production of [H2S]i is increased to levels high enough to affect the function of ion channels or other cellular targets such as mitochondrial cytochrome oxidase in a persistent manner. Chronic hypoxia is well known to be associated with sensitization of CB sensory response to hypoxia (Chen et al., 2002; Powell, 2007; Prabhakar and Jacono, 2005), and this could involve a sustained elevation of [H2S] in the CB. Sensitization of CB sensory response is also a well-documented phenomenon in pathological states such as sleep apnea, hypertension, heart failure and insulin resistance, and may involve a chronically elevated [H2S] in the CB (Iturriaga et al., 2014; Ribeiro et al., 2013; Schultz and Li, 2007). A recent study showed that inhibition of H2S production with PAG restored normal breathing and improved autonomic function in experimental heart failure induced in rats (Del Rio et al., 2013). Thus, a persistent elevation of [H2S] during chronic hypoxia could be an important cause of CB sensitization. It would be interesting to test whether a persistent elevation of [H2S] affects TASK function and [Ca2+]i.
Highlights.
H2S inhibited TASK activity, caused depolarization and elevated intracellular [Ca]i in carotid body glomus cells.
Inhibitors of cystathionine-β-synthase and cystathionine-γ-lyase abolished the production of H2S in glomus cells.
However, the inhibitors of these enzymes failed to block hypoxia-induced inhibition of TASK activity, depolarization or the elevation of intracellular [Ca]i.
Thus, our results show that the level of endogenously produced H2S by hypoxia is not sufficiently high to cause glomus cell excitation, and that under normal conditions, a signal other than H2S mediates the hypoxia-induced excitation of glomus cells.
Acknowledgments
We thank Drs. Christopher J. Chang, Alexander R. Lippert and Vivian S. Lin for providing SF7-AM. We thank James O. Hogan for preparing glomus cells from rat carotid body. This work was funded by grant award to D. Kim (NIH111497).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Donghee Kim, Email: Donghee.kim@rosalindfranklin.edu.
John L. Carroll, Email: carrolljohnl@uams.edu.
References
- Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A. Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE) Br J Pharmacol. 2013;169:922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. TASK-like potassium channels and oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2007;157:55–64. doi: 10.1016/j.resp.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012;463:743–754. doi: 10.1007/s00424-012-1089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JL, Kim I. Carotid chemoreceptor “resetting” revisited. Respir Physiol Neurobiol. 2013;185:30–43. doi: 10.1016/j.resp.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, He L, Dinger B, Stensaas L, Fidone S. Role of endothelin and endothelin A-type receptor in adaptation of the carotid body to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1314–1323. doi: 10.1152/ajplung.00454.2001. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Marcus NJ, Schultz HD. Inhibition of hydrogen sulfide restores normal breathing stability and improves autonomic control during experimental heart failure. J Appl Physiol (1985) 2013;114:1141–1150. doi: 10.1152/japplphysiol.01503.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombkowski RA, Doellman MM, Head SK, Olson KR. Hydrogen sulfide mediates hypoxia-induced relaxation of trout urinary bladder smooth muscle. J Exp Biol. 2006;209:3234–3240. doi: 10.1242/jeb.02376. [DOI] [PubMed] [Google Scholar]
- Elsey DJ, Fowkes RC, Baxter GF. L-cysteine stimulates hydrogen sulfide synthesis in myocardium associated with attenuation of ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther. 2010;15:53–59. doi: 10.1177/1074248409357743. [DOI] [PubMed] [Google Scholar]
- Fernandes VS, Ribeiro AS, Martinez P, Lopez-Oliva ME, Barahona MV, Orensanz LM, Martinez-Saenz A, Recio P, Benedito S, Bustamante S, Garcia-Sacristan A, Prieto D, Hernandez M. Hydrogen sulfide plays a key role in the inhibitory neurotransmission to the pig intravesical ureter. PLoS One. 2014;9:e113580. doi: 10.1371/journal.pone.0113580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald RS, Shirahata M, Chang I, Kostuk E, Kiihl S. The impact of hydrogen sulfide (H(2)S) on neurotransmitter release from the cat carotid body. Respir Physiol Neurobiol. 2011;176:80–89. doi: 10.1016/j.resp.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Haouzi P, Bell H, Philmon M. Hydrogen sulfide oxidation and the arterial chemoreflex: effect of methemoglobin. Respir Physiol Neurobiol. 2011a;177:273–283. doi: 10.1016/j.resp.2011.04.025. [DOI] [PubMed] [Google Scholar]
- Haouzi P, Bell H, Van de Louw A. Hypoxia-induced arterial chemoreceptor stimulation and hydrogen sulfide: too much or too little? Respir Physiol Neurobiol. 2011b;179:97–102. doi: 10.1016/j.resp.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Iturriaga R, Andrade DC, Del Rio R. Enhanced carotid body chemosensory activity and the cardiovascular alterations induced by intermittent hypoxia. Front Physiol. 2014;5:468. doi: 10.3389/fphys.2014.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Kimura Y, Kimura H, Niki I. L-cysteine inhibits insulin release from the pancreatic beta-cell: possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes. 2006;55:1391–1397. doi: 10.2337/db05-1082. [DOI] [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I, Carroll JL. Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. J Physiol. 2009;587:2963–2975. doi: 10.1113/jphysiol.2009.171181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal. 2010;12:1179–1189. doi: 10.1089/ars.2009.2926. [DOI] [PubMed] [Google Scholar]
- Lin VS, Chang CJ. Fluorescent probes for sensing and imaging biological hydrogen sulfide. Curr Opin Chem Biol. 2012;16:595–601. doi: 10.1016/j.cbpa.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin VS, Lippert AR, Chang CJ. Cell-trappable fluorescent probes for endogenous hydrogen sulfide signaling and imaging H2O2-dependent H2S production. Proc Natl Acad Sci U S A. 2013;110:7131–7135. doi: 10.1073/pnas.1302193110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert AR, New EJ, Chang CJ. Reaction-based fluorescent probes for selective imaging of hydrogen sulfide in living cells. J Am Chem Soc. 2011;133:10078–10080. doi: 10.1021/ja203661j. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, del Toro R, Levitsky KL, Chiara MD, Ortega-Saenz P. Regulation of oxygen sensing by ion channels. J Appl Physiol. 2004;96:1187–1195. doi: 10.1152/japplphysiol.00929.2003. [DOI] [PubMed] [Google Scholar]
- Makarenko VV, Nanduri J, Raghuraman G, Fox AP, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am J Physiol Cell Physiol. 2012;303:C916–923. doi: 10.1152/ajpcell.00100.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y, Nagahata Y, Ishikawa M, Hattori K, Takenouchi T, Takahashi T, Ishii I, Matsubara K, Kabe Y, Uchiyama S, Nagata E, Gadalla MM, Snyder SH, Suematsu M. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc Natl Acad Sci U S A. 2012;109:1293–1298. doi: 10.1073/pnas.1119658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR. Hydrogen sulfide and oxygen sensing: implications in cardiorespiratory control. J Exp Biol. 2008;211:2727–2734. doi: 10.1242/jeb.010066. [DOI] [PubMed] [Google Scholar]
- Olson KR, Dombkowski RA, Russell MJ, Doellman MM, Head SK, Whitfield NL, Madden JA. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol. 2006;209:4011–4023. doi: 10.1242/jeb.02480. [DOI] [PubMed] [Google Scholar]
- Olson KR, Healy MJ, Qin Z, Skovgaard N, Vulesevic B, Duff DW, Whitfield NL, Yang G, Wang R, Perry SF. Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am J Physiol Regul Integr Comp Physiol. 2008;295:R669–680. doi: 10.1152/ajpregu.00807.2007. [DOI] [PubMed] [Google Scholar]
- Otsubo T, Kostuk EW, Balbir A, Fujii K, Shirahata M. Differential Expression of Large-Conductance Ca-Activated K Channels in the Carotid Body between DBA/2J and A/J Strains of Mice. Front Cell Neurosci. 2011;5(11–18):19. doi: 10.3389/fncel.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardal R, Ludewig U, Garcia-Hirschfeld J, Lopez-Barneo J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci U S A. 2000;97:2361–2366. doi: 10.1073/pnas.030522297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteus CS, Abdallah SJ, Pollack J, Kumai Y, Kwong RW, Yew HM, Milsom WK, Perry SF. The role of hydrogen sulphide in the control of breathing in hypoxic zebrafish (Danio rerio) J Physiol. 2014;592:3075–3088. doi: 10.1113/jphysiol.2014.271098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell FL. The influence of chronic hypoxia upon chemoreception. Respir Physiol Neurobiol. 2007;157:154–161. doi: 10.1016/j.resp.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Jacono FJ. Cellular and molecular mechanisms associated with carotid body adaptations to chronic hypoxia. High Alt Med Biol. 2005;6:112–120. doi: 10.1089/ham.2005.6.112. [DOI] [PubMed] [Google Scholar]
- Prieto-Lloret J, Shaifta Y, Ward JP, Aaronson PI. Hypoxic pulmonary vasoconstriction in isolated rat pulmonary arteries is not inhibited by antagonists of H2S-synthesizing pathways. J Physiol. 2014 doi: 10.1113/jphysiol.2014.277046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MJ, Sacramento JF, Gonzalez C, Guarino MP, Monteiro EC, Conde SV. Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes. 2013;62:2905–2916. doi: 10.2337/db12-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz HD, Li YL. Carotid body function in heart failure. Respir Physiol Neurobiol. 2007;157:171–185. doi: 10.1016/j.resp.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D, Kemp PJ. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol. 2010;172:169–178. doi: 10.1016/j.resp.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Varas R, Wyatt CN, Buckler KJ. Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. J Physiol. 2007;583:521–536. doi: 10.1113/jphysiol.2007.135657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Williams BA, Buckler KJ. Biophysical properties and metabolic regulation of a TASK-like potassium channel in rat carotid body type 1 cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L221–230. doi: 10.1152/ajplung.00010.2003. [DOI] [PubMed] [Google Scholar]